Atomsk
Le couteau suisse pour les simulations atomiques

Le couteau suisse pour les simulations atomiques




Dans ce tutoriel vous allez apprendre à générer des mailles élémentaires pour différents types de cristaux avec Atomsk. Vous allez aussi apprendre à générer des fichiers pour différents logiciels de visualisation. Pour la visualisation, il est recommandé d'installer OVITO, Atomeye, XCrySDen, et/ou VESTA.
▶ Pour plus d'information, référez-vous à la page de documentation correspondante.
Générer une maille élémentaire peut être utile pour réaliser des illustrations, par exemple pour expliquer les rudiments de la cristallographie à des étudiants ou au grand public. Dans le cadre de simulations à l'échelle atomique, c'est souvent le premier pas avant de construire des systèmes plus complexes. Atomsk peut être utilisé pour générer certains types de mailles élémentaires, grâce au mode "--create".
En plus de ce tutoriel, il est recommandé de lire la page de documentation du mode "--create".
De nombreux métaux, comme l'aluminium, le cuivre, ou encore le nickel, cristallisent dans un réseau cfc.
Commençons avec l'aluminium (symbole Al). L'aluminium est un métal cfc avec un paramètre de maille a=4.046 Å à température ambiante. Pour générer sa maille élémentaire, Atomsk peut être exécuté de la façon suivante :
atomsk --create fcc 4.046 Al aluminium.cfg
ⓘ Ce paramètre de maille est donné comme exemple, et doit être ajusté en fonction du type de simulation que vous voulez effectuer (DFT, potentiel interatomique, etc.).
Avec cette commande, Atomsk va écrire les positions des atomes dans le fichier de sortie "aluminium.cfg". Il s'agit d'un fichier texte, et si vous l'ouvrez avec un éditeur de texte (comme Notepad) il ressemble à ceci :
Number of particles = 4
# Fcc Al oriented X=[100] Y=[010] Z=[001].
A = 1.000000000 Angstrom (basic length-scale)
H0(1,1) = 4.04600000
H0(1,2) = 0.00000000
H0(1,3) = 0.00000000
H0(2,1) = 0.00000000
H0(2,2) = 4.04600000
H0(2,3) = 0.00000000
H0(3,1) = 0.00000000
H0(3,2) = 0.00000000
H0(3,3) = 4.04600000
.NO_VELOCITY.
entry_count = 3
26.9815
Al
0.00000000 0.00000000 0.00000000
0.50000000 0.50000000 0.00000000
0.00000000 0.50000000 0.50000000
0.50000000 0.00000000 0.50000000
Ce fichier respecte le format CFG, tel que défini par Ju Li pour son logiciel de visualisation Atomeye. La première ligne indique que le système contient 4 atomes. La deuxième ligne (commençant par #) est un commentaire qui est généré automatiquement par Atomsk. S'en suivent les vecteurs de boîte (lignes commençant par H0), suivis par des mots-clés, et enfin la masse et le symbole chimique des atomes, et leurs positions. Notez que dans ce format, les positions sont données en coordonnées réduites, c'est-à-dire comme une fraction des vecteurs de boîte.
Un tel fichier au format CFG peut être ouvert avec OVITO, un logiciel de visualisation et d'analyse conçu par Alexander Stukowski et co-auteurs, et qui permet de visualiser la maille en 3-D :
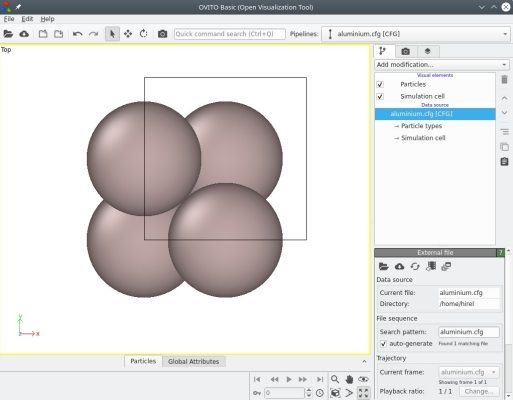
Nous pouvons voir que OVITO affiche la base cartésienne (flèches colorées estampillées X, Y, Z en bas à gauche), les contours de la maille (boîte noire), ainsi que les quatre atomes comme attendu (sphères grises).
Maintenant, ouvrons exactement le même fichier "aluminium.cfg" avec un autre logiciel de visualisation, Atomeye :
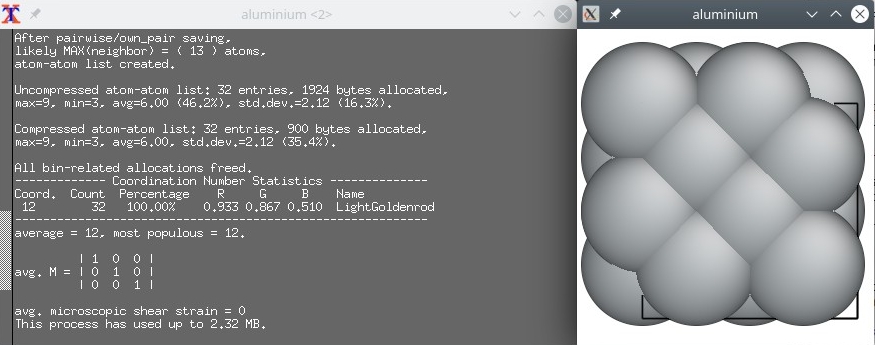
Ici nous voyons un comportement qui est spécifique à Atomeye. Le système est très petit (c'est seulement une maille élémentaire), alors Atomeye le duplique automatiquement dans les trois directions. Dans son terminal il affiche qu'il y a "32 atomes" dans le système. C'est juste un artefact de la visualisation, car nous savons qu'en réalité notre système ne contient que 4 atomes. C'est une leçon importante à retenir : le nombre d'atomes affichés par un logiciel de visualisation ne correspond pas toujours au nombre d'atomes existant dans le fichier de données.
Pour changer, construisons une maille de cuivre. Le cuivre (symbole Cu) a un réseau cfc comme l'aluminium précédemment, dont la maille contiendra 4 atomes également, mais son paramètre de maille est différent : a=3.597 Å. Cette fois, écrivons les données à la fois au format CFG, et au format XSF :
atomsk --create fcc 3.597 Cu copper.cfg xsf
Notez que, en plus du fichier nommé "copper.cfg", nous avons ajouté le mot-clé "xsf" à la fin de la ligne de commandes. Cette fois, Atomsk va générer deux fichiers de sortie : "copper.cfg" et "copper.xsf". Ouvrons le premier fichier, "copper.cfg", avec OVITO :
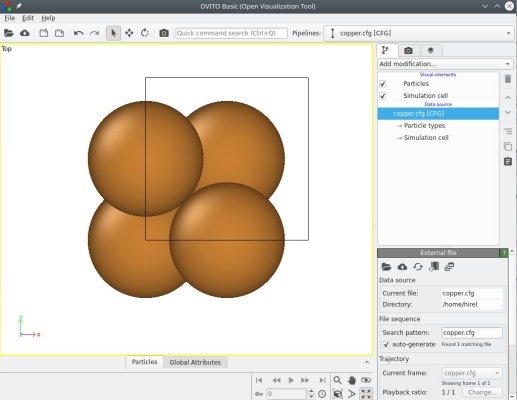
Pas grand chose de nouveau ici, nous voyons à peu près la même chose que dans le cas de l'aluminium. OVITO utilise des couleurs différentes pour les atomes de différentes espèces chimiques, gris pour l'aluminium, orange pour le cuivre.
Maintenant essayons de visualiser le deuxième fichier, "copper.xsf" -qui contient bien sûr aussi 4 atomes. OVITO peut aussi importer les fichiers XSF, vous pouvez donc vérifier que OVITO affiche la même chose à partir des deux fichiers. Essayons plutôt un autre logiciel de visualisation. Le format XSF a été initialement conçu par Anton Kokalj pour son logiciel XCrySDen, ouvrons donc notre fichier "copper.xsf" avec XCrySDen :
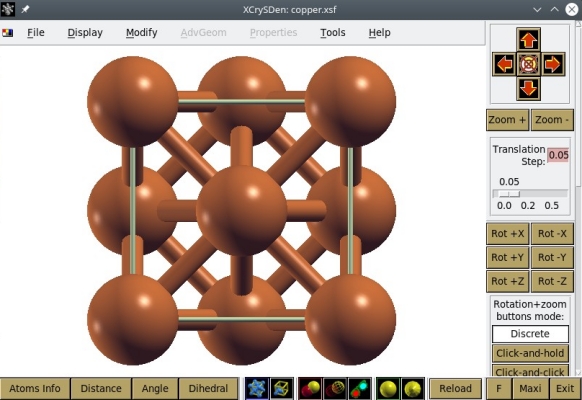
Alternativement, les fichiers XSF peuvent aussi être ouverts avec VESTA, comme illustré ci-dessous :
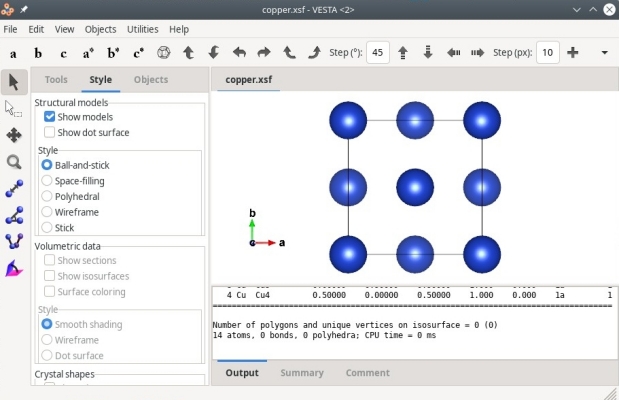
Comme précédemment, nous savons qu'il n'y a que 4 atomes dans notre fichier, mais XCrySDen ou VESTA en affichent plus que cela. C'est parce que ces logiciels appliquent automatiquement des conditions aux limites périodiques, et affichent les répliques des atomes si elles se trouvent dans la boîte. Par exemple, l'atome de coordonnées (0,0,0) apparaît aussi à chaque coin de la boîte à cause de cette périodicité. C'est pourquoi ces logiciels affichent une boîte contenant 14 atomes. Encore une fois, retenez que ce que vous voyez dans un logiciel de visualisation ne correspond pas forcément au contenu de votre fichier de données.
Comme deuxième exemple, tournons-nous vers un deuxième type de réseau, le réseau cubique centré. C'est le réseau cristallin de nombreux métaux de transition, comme le fer (Fe) ou le tungstène (W).
Évidemment, dans la ligne de commande nous devons remplacer "fcc" par "bcc". Par exemple, pour le fer avec un paramètre de maille de 2.856 Å, nous pouvons utiliser :
atomsk --create bcc 2.856 Fe xsf cfg
Dans cette commande, nous n'avons pas spécifié de nom de fichier de sortie. Seuls deux formats sont donnés : "xsf" et "cfg". Dans cette situation, Atomsk utilise le nom de l'élément (ici "Fe") pour générer les noms de fichiers. Ainsi, deux fichiers seront écrits par Atomsk : "Fe.cfg" qui peut être visualisé avec OVITO (ou Atomeye), et "Fe.xsf" qui peut être ouvert avec VESTA (ou XCrySDen).
De façon similaire, pour générer une maille élémentaire de tungstène avec le paramètre de maille 3.155 Å:
atomsk --create bcc 3.155 W xsf
Là encore, Atomsk utilisera le nom de l'élément ("W") pour générer le nom du fichier de sortie, "W.xsf".
La maille élémentaire cubique centrée contient deux atomes. Les fichiers que nous venons de générer, "Fe.xsf" et "W.xsf", peuvent être ouverts avec VESTA :
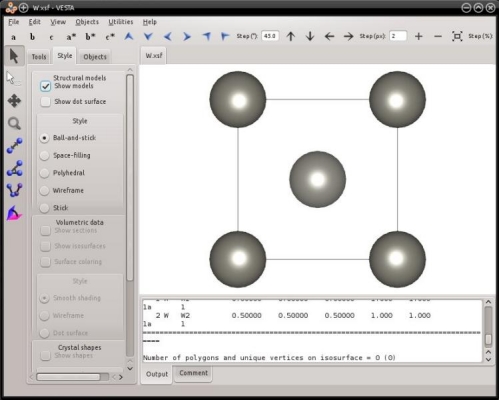
Comme expliqué précédemment, VESTA suppose le système périodique, et affiche les répliques périodiques des atomes.
Les cristaux précédents sont assez simples et ne contiennent qu'un seul type d'atomes. Certains cristaux peuvent renfermer des atomes de plusieurs espèces chimiques différentes, comme c'est le cas du sel de table : le chlorure de sodium (NaCl) avec le réseau dit "rock-salt".
Atomsk peut générer ce type de maille avec le mot-clé "rocksalt". Après le paramètre de maille (5.64 Å pour NaCl), deux symboles chimiques doivent être donnés :
atomsk --create rocksalt 5.64 Na Cl xsf
ⓘ Par défaut, Atomsk ne génère que les positions atomiques, et n'attribue pas de charges électriques aux ions. Pour définir des charges électriques, rendez-vous sur ce tutoriel.
Avec ces instructions, Atomsk va construire une maille de NaCl, et l'écrire dans le fichier "NaCl.xsf", qui peut être visualisé avec VESTA :
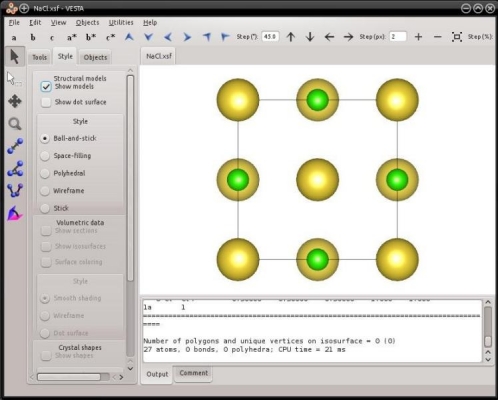
Le mode "--create" ne se limite pas aux mailles cubiques. Il est aussi possible de générer des mailles haxagonales, comme l'hexagonal compact (hcp), wurtzite, ou encore le graphite.
Commençons par créer une maille de magnésium hcp. Deux paramètres de maille, a et c, doivent être données :
atomsk --create hcp 3.21 5.213 Mg Mg.xsf
La visualization (ici avec VESTA) montre qu'en effet, une maille de magnésium hcp a été créée :
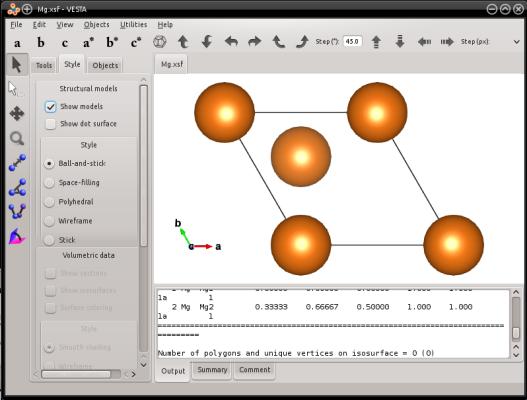
Si vous souhaitez obtenir une maille équivalente orthogonale (mais qui respecte toujours la périodicité du réseau hexagonal), vous pouvez utiliser l'option "-orthogonal-cell". Cette dernière recherche des combinaisons linéaires des vecteurs de boîte, qui donnent une nouvelle boîte orthogonale, puis elle remplit cette nouvelle boîte avec des atomes. Dans le cas du magnésium hcp, vous pouvez utiliser :
atomsk --create hcp 3.21 5.213 Mg -orthogonal-cell Mg.xsf
Si vous visualisez ce système, vous verrez que c'est une maille orthogonale de magnésium hcp :
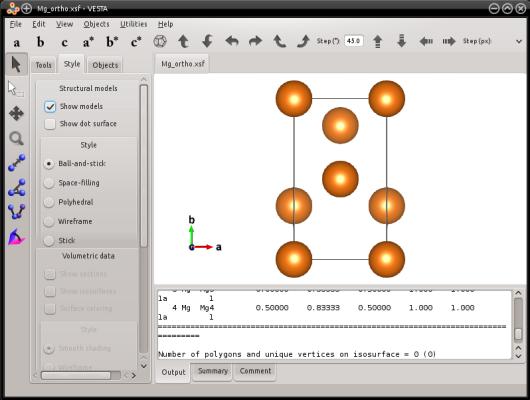
De façon similaire, Atomsk permet aussi de créer une maille de graphite :
atomsk --create graphite 3.21 5.213 C graphite.xsf
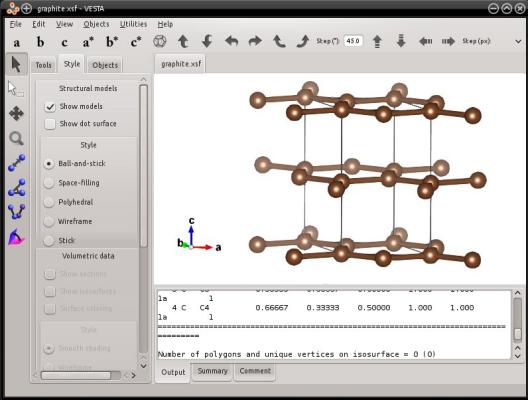
Ici, VESTA affiche des liaisons, et affiche aussi des atomes de carbone qui sont en dehors de la boîte. Mais le fichier texte ne contient en réalité que les quatre atomes de la maille de graphite, comme vous pouvez le vérifier en l'ouvrant avec un éditeur de texte.
Atomsk peut générer des mailles pour différents réseaux cubiques, tétragonaux, ou hexagonaux. Veuillez lire la page de documentation pour savoir quels types de réseaux sont supportés.
Et si je veux créer une maille qui n'est pas supportée par Atomsk ? Dans ce cas, le mieux est d'obtenir un fichier décrivant la structure depuis une autre source. En particulier, de nombreuses structures sont décrites dans des fichiers au format CIF, qui peuvent être obtenues depuis des bases de données en ligne comme Crystallography Open Database ou American Mineralogist database. Vous pouvez aussi entrer le nom du matériau que vous recherchez suivi du mo-clé "CIF" dans votre moteur de recherche préféré. Ce tutoriel explique comment importer des fichiers CIF avec Atomsk.
Alternativement, si vous connaissez le groupe d'espace et les positions de Wickoff du composé, vous pouvez utiliser un logiciel dédié pour construire la maille élémentaire. Par exemple, VESTA contient un outil capable de construire des structures atomiques (ouvrez VESTA, allez dans File → New structure).
Enfin, la plupart du temps une maille élémentaire ne contient qu'un petit nombre d'atomes. Vous pouvez écrire les positions des atomes dans un fichier texte, avec les vecteurs de boîte. Assurez-vous de les écrire dans un format que Atomsk peut lire, comme le format XYZ (voir la la liste des formats pris en charge). Ensuite, Atomsk peut lire votre fichier et manipuler les données.
Souvent, il est nécessaire de dupliquer la maille élémentaire pour obtenir un cristal plus grand, appelé "super-cellule". Cela peut être réalisé avec l'option "-duplicate". Cette option peut être utilisée en combinaison du mode "--create", par exemple la commande suivante va créer une maille de tungstène, puis la dupliquer 3 fois suivant X, 4 fois suivant Y, et 10 fois suivant Z :
atomsk --create bcc 3.155 W -duplicate 3 4 10 xsf
Ou, si vous avez déjà un fichier contenant une maille élémentaire (ou n'importe quel système atomique), alors vous pouvez la lire puis la dupliquer :
atomsk initial.xsf -duplicate 3 4 10 final.cfg
ⓘ Ces valeurs sont à considérer comme des "facteurs de multiplication" suivant chaque direction. Ainsi, une valeur de 1 signifie que le système est multiplié par 1, c'est-à-dire que sa dimension ne change pas suivant la direction donnée ("-duplicate 1 1 1" ne changerait rien du tout au système). Une valeur de 0 (zéro) est automatiquement remplacée par 1, parce que multiplier par zéro supprimerait complètement le système. Les valeurs négatives signifient que le système est retourné suivant la direction donnée, avant d'être multiplié.
La duplication utilise toujours les vecteurs de boîte comme vecteurs de translation, de façon à préserver la périodicité du cristal. Ainsi, si vos vecteurs de boîte ne sont pas orthogonaux ou s'ils ne sont pas alignés avec les axes cartésiens, ne vous en faites pas Atomsk n'utilise pas les directions cartésiennes pour dupliquer, mais les vecteurs de la boîte donnée en entrée, peu importe ce qu'ils sont.
Comme vous pouvez le voir avec les exemples précédents, il est très simple de générer des fichiers pour la visualisation avec Atomsk. Si vous avez un logiciel de visualisation préféré, vous pouvez adapter les tutoriels suivants pour générer des fichiers dans le format qui vous convient le mieux. Par exemple si vous êtes familier avec Atomeye ou OVITO, alors écrivez vos fichiers au format CFG. Si vous êtes plus familier avec VESTA ou XCrySDen, alors utilisez le format XSF.
Si vous souhaitez choisir l'orientation cristalline de la maille, veuillez vous rendre au tutoriel suivant.
Maintenant utilisez votre imagination ! Vous pouvez changer les éléments chimiques à l'envi. Vous pouvez aussi lire la documentation du mode --create et générer différentes mailles cristallines, ou même des nanotubes.