Atomsk
Le couteau suisse pour les simulations atomiques

Le couteau suisse pour les simulations atomiques




Ce tutoriel explique comment Atomsk lit et traite les fichiers CIF.
Les tutoriels précédents ont illustré comment créer des systèmes atomiques avec le mode "--create". Cependant ce mode ne supporte qu'un nombre limité de réseaux cristallins, et devient complètement inutile si vous voulez modéliser des structures plus complexes.
Le format des fichiers d'informations cristallographiques ou Crystallographic Information File (CIF) est un format standard, conçu par l'Union Internationale de Cristallographie. C'est un format très puissant et très utilisé. Des fichiers CIF pour de nombreux composés peuvent être téléchargés depuis la base de données de structures cristallines de American Mineralogist, ou depuis la base de données ouverte de cristallographie, par exemple.
Un fichier au format CIF peut contenir les positions des atomes, ainsi que le groupe de symétrie auquel le composé appartient. C'est donc un format un peu particulier : toutes les positions des atomes ne sont pas explicitement écrites dans un fichier CIF, mais elles peuvent être déduites en appliquant les symétries du groupe d'espace indiqué. C'est exactement ce que fait Atomsk lorsqu'il lit un fichier CIF.
À titre d'exemple, téléchargez le fichier CIF correspondant au titanate de strontium (SrTiO3) avec la structure perovskite, depuis la base de données ouverte de cristallographie :
Fichier CIF pour SrTiO3 depuis la base de données ouverte
Le nom du fichier est "9002806.cif". Si vous l'ouvrez avec un éditeur de texte, vous verrez qu'il contient plusieurs informations :
Comme expliqué précédemment, un fichier CIF ne contient pas les positions de tous les atomes dans la boîte. En tout cas, pas explicitement. Ici, le fichier ne spécifie que les positions de quatre atomes (Sr, Ti, et deux atomes d'oxygène), alors que la formule chimique indique "SrTiO3". Toutefois, le fichier CIF contient le groupe d'espace et les opérations de symétrie correspondant à ce composé. En appliquant ces opérations aux atomes, il est possible de trouver tous les sites équivalents, et de remplir la boîte avec des atomes.
Pour poursuivre cet exemple, convertissons ce fichier CIF au format VESTA pour le visualiser :
atomsk 9002806.cif vesta
Vous pouvez remarquer que Atomsk indique : "Application des opérations de symétrie". Il est important de réaliser que, lorsqu'il lit un fichier CIF, Atomsk applique les opérations de symétrie immédiatement. Dit autrement, il trouve les positions équivalentes pour tous les atomes dans la boîte.
La commande précédente génère un fichier nommé "9002806.vesta", qui contient les 20 atomes de la boîte. La visualisation avec VESTA le confirme :
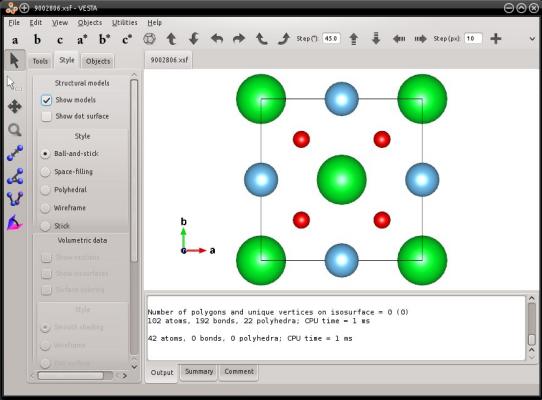
Après avoir lu le fichier CIF, Atomsk perd toute information concernant le groupe d'espace : toutes les positions atomiques sont désormais connues, et le groupe d'espace devient juste P1. C'est pourquoi dans le fichier VESTA, Atomsk écrit comme groupe d'espace "P1".
Parfois, un fichier CIF peut contenir des informations sur des occupations partielles. Cela signifie que le même site cristallographique peut être occupé par des atomes d'espèces chimiques différentes. Pour l'exemple, téléchargez un autre fichier depuis la base de données :
Fichier CIF pour Ca0.35Sr0.65TiO3 depuis la base de données ouverte
Ce composé est similaire au précédent, sauf que cette fois, des atomes de calcium (Ca) peuvent substituer des atomes de strontium (Sr). Si vous ouvrez ce fichier CIF ("9002807.cif"), vous verrez que la section portant sur les atomes contient une colonne supplémentaire, nommée "_atom_site_occupancy". Tous les atomes ont une occupation de 1 (ce qui veut dire qu'ils occupent 100% de leurs sites), sauf Ca et Sr qui ont des occupations de 0.35 et 0.65 respectivement. Vous pouvez aussi remarquer que les atomes de Ca et Sr sont situés exactement à la même position.
Importons ce fichier avec Atomsk, et convertissons-le au format VESTA pour visualisation :
atomsk 9002807.cif vesta
Dans cet exemple, Atomsk lit l'information sur les occupations partielles depuis le fichier CIF, et peut donc les écrire dans le fichier de sortie au format VESTA. Visualisons-le :
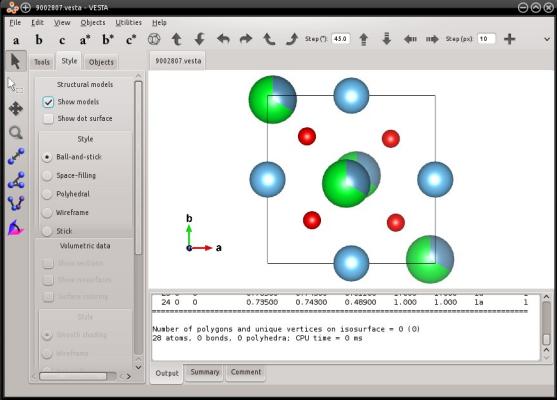
VESTA affiche les occupations partielles avec des sphères partiellement remplies, confirmant que cette information a bien été conservée par Atomsk pendant la conversion.
Maintenant, supposons que nous voulions effectuer une simulation avec LAMMPS. Convertissons le fichier CIF directement au format de fichier d'entrée de LAMMPS :
atomsk 9002807.cif lammps
Dans ce cas, Atomsk va afficher un avertissement :
/!\ ALERTE : les données d'entrée contiennent des occupations partielles,
que certains formats de sortie ne supportent pas.
Certains atomes risquent de se chevaucher dans les fichiers de sortie.
En effet, il n'est pas possible d'écrire les informations sur les occupations partielles dans un fichier au format LAMMPS, parce que ce format ne le supporte pas. Cette information est donc perdue, et on se retrouve avec un système atomique dans lequel certains atomes partagent exactement la même position, ce qui n'est pas physique. Dans un tel cas, c'est à l'utilisateur (vous-même) de décider ce qu'il veut faire des atomes qui se chevauchent.
En lisant des fichiers CIF, Atomsk enregistre les informations sur les occupations partielles. Puis, si c'est possible, il les écrit dans le(s) fichier(s) de sortie. Quand ce n'est pas possible, il affiche le message d'avertissement ci-dessus.
Dans Atomsk, l'option "--create" permet de choisir l'orientation d'un cristal au moment de sa création. Toutefois ce n'est pas possible lorsqu'un fichier d'entrée est fourni (comme un fichier CIF), il faut donc recourir à d'autres méthodes pour changer l'orientation du cristal.
Utilisons le fichier CIF pour SrTiO3 ci-dessus comme exemple (le fichier "9002806.cif"). D'abord nous devons déterminer l'orientation cristalline de départ. Il s'avère que dans ce fichier, le cristal est décrit dans l'orientation X=[110], Y=[110], Z=[001] (cela peut être déduit des symétries de ce cristal).
Maintenant, si nous voulons obtenir une autre orientation, nous devons appliquer une rotation. Supposons que nous voulions X=[100], Y=[010], Z=[001]. Puisque ce cristal est cubique, nous pouvons utiliser l'option "-orient", où l'on doit fournir les orientations cristallines initiale et finale :
atomsk 9002806.cif -orient [110] [-110] [001] [100] [010] [001] SrTiO3_rotated.cfg
De façon alternative, on peut démontrer qu'on obtient le même résultat en effectuant une rotation de 45° autour de l'axe Z, ce qui peut être fait avec l'option "-rotate" :
atomsk 9002806.cif -rotate Z 45 SrTiO3_rotated.cfg
Les deux commandes produisent le même résultat, comme on peut le vérifier avec OVITO :
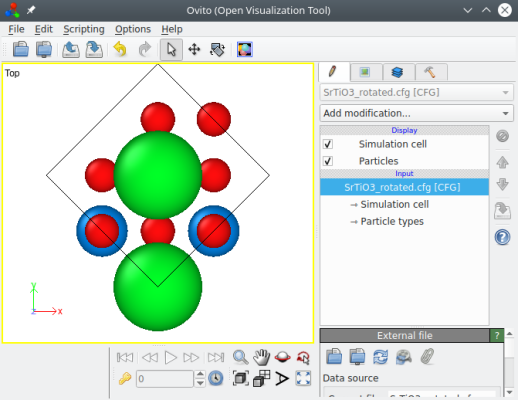
Maintenant nous avons la bonne orientation cristallographie, mais la boîte est tournée, ses vecteurs ne sont pas alignés avec les axes cartésiens. La plupart du temps, une boîte orthogonale est plus pratique. Nous pouvons l'obtenir grâce à l'option "-orthogonal-cell":
atomsk 9002806.cif -rotate Z 45 -orthogonal-cell SrTiO3_rotated.cfg
Cette fois le système est tourné, et nous avons une boîte orthogonale "correct" :
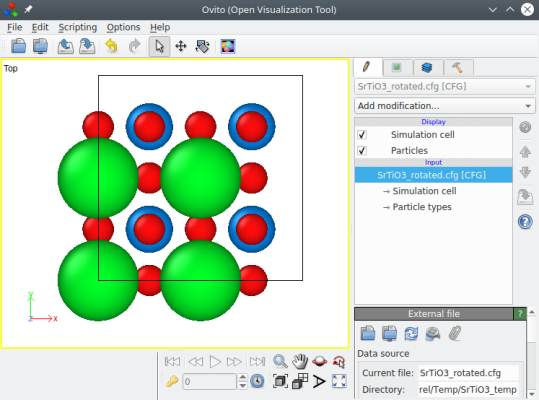
Pour résumer pour orienter une maille lue depuis un fichier source (par exemple un fichier CIF), vous devez d'abord déterminer l'orientation ou l'angle de rotation nécessaire, puis utiliser l'option "-orthogonal-cell".
Atomsk ne peut écrire que des fichiers CIF simples. Il suppose toujours que le groupe d'espace est P1, et écrit explicitement toutes les positions des atomes dans le fichier CIF.