Atomsk
Le couteau suisse pour les simulations atomiques

Le couteau suisse pour les simulations atomiques




Ce tutoriel explique comment définir, ajouter ou modifier des propriétés auxiliaires des atomes avec Atomsk.
▶ Pour plus d'informations, référez-vous à la page de documentation correspondante.
L'information minimale pour définir un système atomique est l'espèce chimique des atomes et leurs positions. Cependant il est souvent utile de définir des informations additionnelles. Dans Atomsk, cela peut être réalisé avec l'option "-properties", qui lit des propriétés depuis un fichier texte.
Atomsk distingue deux types de propriétés celles qui prennent une valeur différente pour chaque atome, appelées "propriétés par atome" ou "propriétés auxiliaires" et les propriétés globales du système.
Une chose importante à comprendre, est que tous les formats de fichiers n'implémentent pas les propriétés auxiliaires de la même façon.
Donc, si vous souhaitez conserver les propriétés auxiliaires lorsque vous convertissez des fichiers, assurez-vous que le fichier de sortie que vous choisissez supporte bien les propriétés que vous avez définies pour votre système. Sinon, si le format de fichier ne le supporte pas, alors Atomsk ne peut pas écrire ces propriétés et leurs données seront simplement perdues. Pour savoir quelles propriétés auxiliaires sont supportées par quels formats , veuillez vous référer à la liste des formats supportés par Atomsk.
Lorsqu'il lit un fichier d'entrée, Atomsk essaye de lire les propriétés auxiliaires aussi. Encore une fois, la liste des formats indique quelles propriétés peuvent être importées pour chaque format. En convertissant d'un format vers un autre, Atomsk essaye de transférer les propriétés lorsque c'est possible. Cependant, si le format de sortie choisi ne supporte pas les mêmes propriétés que le fichier d'entrée, elles ne peuvent pas être transférées et sont perdues.
Dans la suite, le format CFG sera largement utilisé, parce qu'il permet d'enregistrer n'importe quelle propriété, et peut facilement être visualisé avec Atomeye ou OVITO.
Commençons avec une propriété qui prend une valeur pour chaque atome. À titre d'exemple, supposons que nous souhaitons construire un cristal ionique, pour lequel la charge électrique portée par chaque ion doit être définie. Créons un cristal de chlorure de sodium NaCl, comme nous l'avons fait dans un précédent tutoriel :
atomsk --create rocksalt 5.64 Na Cl cfg
Maintenant si vous ouvrez le fichier "NaCl.cfg" avec un éditeur de texte, vous verrez que les atomes n'ont pas de charge électrique. En effet, par défaut Atomsk ne génère que les positions des atomes, et il n'a aucune idée si le cristal est ionique ou pas, ou quelles devraient être les charges. Il appartient donc à l'utilisateur de définir les charges ioniques.
Pour y parvenir, écrivons les charges correspondant à chaque espèce chimique dans un fichier texte. Le fichier texte doit contenir le mot-clé "charge", suivi sur chaque ligne, d'un symbole chimique et de la charge correspondante (en unités de charge élémentaire e) :
# Charges pour un cristal de NaCl
charge
Na 1.0
Cl -1.0
Ensuite au moment de générer le cristal de NaCl, utilisons l'option "-properties" pour lire les charges depuis ce fichier :
atomsk --create rocksalt 5.64 Na Cl -properties charge.txt cfg
Maintenant si vous ouvrez le fichier "NaCl.cfg" avec un fichier texte, vous remarquerez qu'il contient une ligne "auxiliary[0] = q", et chaque ligne correspondant à un atome contient quatre colonnes la position x, y, z, et la charge électrique. Vous pouvez aussi ouvrir ce fichier avec OVITO et colorer les atomes avec la propriété "Charge" :
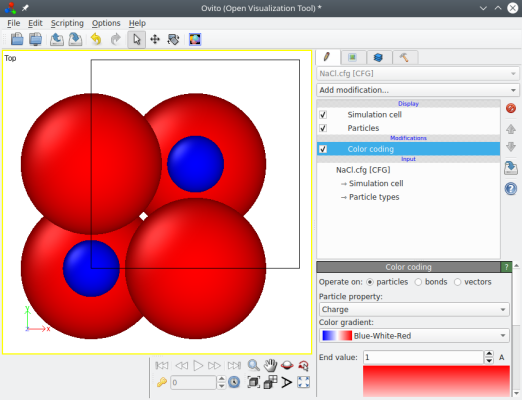
Si vous exécutez la même commande que ci-dessus, en remplaçant "cfg" par "lammps" comme format de sortie, alors Atomsk va produire un fichier de données LAMMPS avec le style "charge" (au lieu du style "atom" par défaut). Vous pouvez en lire plus à ce sujet dans le tutoriel pour LAMMPS.
L'utilisateur peut définir ses propres propriétés auxiliaires, en écrivant dans un fichier texte le mot-clé "auxiliary" suivi du nom de la propriété. Ensuite, chaque ligne doit contenir l'indice d'un atome (un nombre entier) et la valeur de la propriété pour cet atome. Les indices des atomes peuvent apparaître dans n'importe quel ordre. Si un atome n'apparaît pas dans la liste, alors la propriété prend automatiquement la valeur zéro pour cet atome.
À titre d'exemple, supposons que l'énergie potentielle des atomes ait été calculée (par un code de dynamique moléculaire ou tout autre script), et écrite dans un fichier. Comme expliqué précédemment, le mot-clé "auxiliary" doit apparaître suivi du nom de la propriété (ici nous l'appelons "mon_energie"), puis sur les lignes suivantes, un indice et la valeur de l'énergie pour cet atome :
auxiliary mon_energie
2 -20.3
3 -20.4
4 -19.9
6 -19.86
15 -20.4
9 -19.9
atomsk --create rocksalt 5.64 Na Cl -duplicate 2 2 2 -properties energies.txt cfg
ⓘ N'utilisez pas d'espace ni de caractères accentués (é, è, à...) dans le nom de la propriété. Atomsk et d'autres logiciels pourraient avoir du mal à la lire.
Vous pouvez ouvrir le fichier final "NaCl.cfg" avec un éditeur de texte, et vérifier que les énergies y ont bien été écrites. Ce fichier peut aussi être visualisé avec OVITO comme sur la figure ci-dessous : ajoutez "Colour coding" et sélectionnez la propriété "mon_energie".
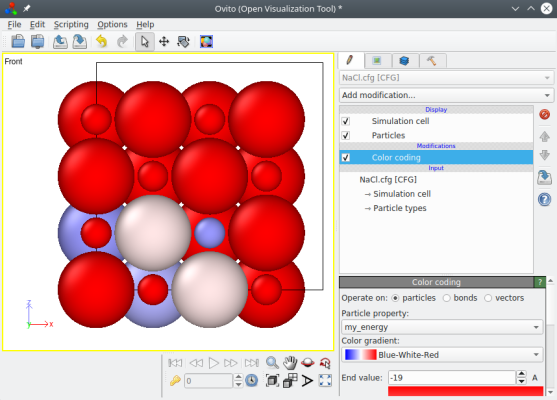
L'option "-properties" est assez versatile, et vous permet d'ajouter n'importe quelle propriété personnalisée à votre système.
Il est aussi possible de définir certaines propriétés qui ne s'appliquent pas à chaque atome individuellement, mais plutôt au système dans son ensemble. C'est le cas par exemple, si vous voulez modifier les vecteurs de boîte sans modifier les positions atomiques. Dans ce cas, écrivez le mot-clé "supercell" suivi des composantes des trois vecteurs de boîte dans un fichier texte :
# Nouveaux vecteurs de boite
supercell
5.64 0.00 0.00
0.00 5.64 0.00
0.00 0.00 5.70
Puis utilisez l'option "-properties" pour l'appliquer :
atomsk NaCl.cfg -properties newbox.txt tetragonal.cfg
Notez que seuls les vecteurs de boîte sont modifiés, tandis que les atomes conservent leurs positions en coordonnées cartésiennes.
Un autre exemple de propriété globale est le tenseur élastique. Pour en savoir plus, vous pouvez vous référer à la documentation, et suivre le tutoriel sur les dislocations en milieu anisotrope.
Après avoir exécuté une simulation ou une analyse, les fichiers peuvent contenir beaucoup de propriétés auxiliaires, dont certaines ne sont plus nécessaire. Atomsk peut supprimer certaines propriétés grâce à l'option "-remove-property" suivie du nom de la propriété à supprimer. Par exemple, si vous voulez vous débarrasser des charges électriques vous pouvez utiliser :
atomsk NaCl.cfg -remove-property q final.cfg
Si vous voulez supprimer toutes les propriétés auxiliaires du système :
atomsk NaCl.cfg -remove-property all final.cfg