Atomsk
Le couteau suisse pour les simulations atomiques

Le couteau suisse pour les simulations atomiques




Ce tutoriel explique comment générer des polycristaux en 3-D ou en 2-D, contenant des grains aléatoires ou avec des orientations cristallines spécifiques.
▶ Pour plus d'information, référez-vous à la page de documentation correspondante.
Le mode "--polycrystal", comme son nom le suggère, est conçu pour générer des polycristaux.
Dans les exemples suivants, une maille élémentaire d'aluminium sera utilisée comme "graine". Une telle maille élémentaire peut être créée avec :
atomsk --create fcc 4.046 Al aluminium.xsf
Atomsk utilise une tesselation de Voronoi pour construire les polycristaux, comme sillustré sur la figure ci-dessous.
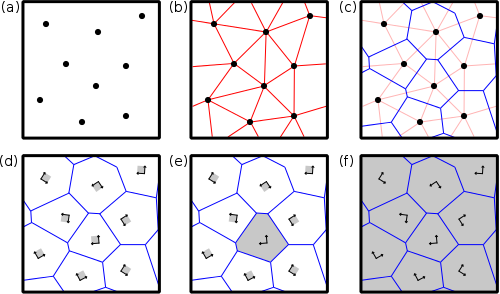
(a) Des nœuds (points noirs) sont introduits à certaines positions données dans la boîte de simulation.
(b) Ces nœuds sont reliés à leurs voisins (lignes rouges), en utilisant des conditions aux limites périodiques.
(c) La normal à chaque ligne rouge est trouvée (lignes bleues). Ces lignes bleues définissent les contours des futurs grains, c'est-à-dire les joints de grains.
(d) Une "graine" atomique (par exemple une maille élémentaire) est placée sur chaque nœud, avec une certaine orientation cristallographique.
(e) Une graine est dupliquée dans les trois directions de l'espace, et les atomes en dehors du grain sont supprimés.
(f) Une fois que toutes les graines ont été dupliquées et coupées dans leurs grains respectifs, on obtient le polycristal final.
Ce schéma illustre la génération d'un polycristal de Voronoi en 2 dimensions, mais peut être généralisé à 3 dimensions.
Commençons par générer un polycristal entièrement aléatoire. Seulement deux informations doivent être fournies à Atomsk : les dimensions de la boîte finale, et le nombre de grains à y placer. Avec ces informations, Atomsk va générer des positions aléatoires pour les nœuds, et des orientations cristallographiques aléatoires pour chaque grain.
D'abord, ces paramètres doivent être écrits dans un fichier texte. Demandons à Atomsk de générer un polycristal de 12 grains, à l'intérieur d'une boîte de taille 200×200×200 Å3 :
box 200 200 200
random 12
Ensuite, exécutons Atomsk en mode "--polycrystal", en utilisant "aluminium.xsf" comme graine, le le fichier texte que nous venons d'écrire :
atomsk --polycrystal aluminium.xsf polycristal.txt final.cfg
Le système final est écrit dans "final.cfg", au format CFG. Atomsk crée automatiquement une nouvelle propriété auxiliaire, appelée "grainID". Cette propriété indique, pour chaque atome, le numéro du grain auquel il appartient (ici de 1 à 12). Le polycristal ainsi généré peut être visualisé, par exemple avec Atomeye (appuyez sur Alt+0 pour visualiser le grainID) :

Atomsk écrit aussi un fichier de données (ici nommé "final.dat") contenant la distribution de tailles de grains. C'est un fichier texte qui peut être ouvert avec un éditeur de texte, ou visualisé sous forme de graphe :
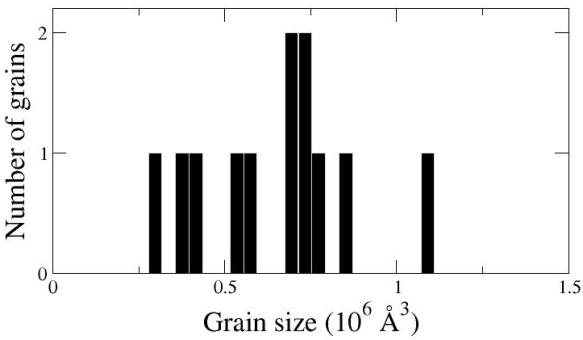
Il est important de noter que "aléatoire" signifie que les nombres générés seront différents à chaque fois que vous exécuterez Atomsk, et donc le polycristal généré sera différent à chaque fois. Si vous exécutez la commande ci-dessus, votre polycristal sera différent de celui montré dans l'image, et il sera aussi différent à chaque fois que vous exécuterez cette même commande.
Lors de la construction d'un polycristal, les mailles élémentaires sont dupliquées à l'intérieur de leur polyèdre de Voronoi en utilisant les coordonnées cartésiennes des atomes. Ainsi, après avoir généré tout le polycristal, certains atomes peuvent se retrouver en-dehors de la boîte. Cela est beaucoup plus clairement visible si vous visualisez le système avec OVITO :
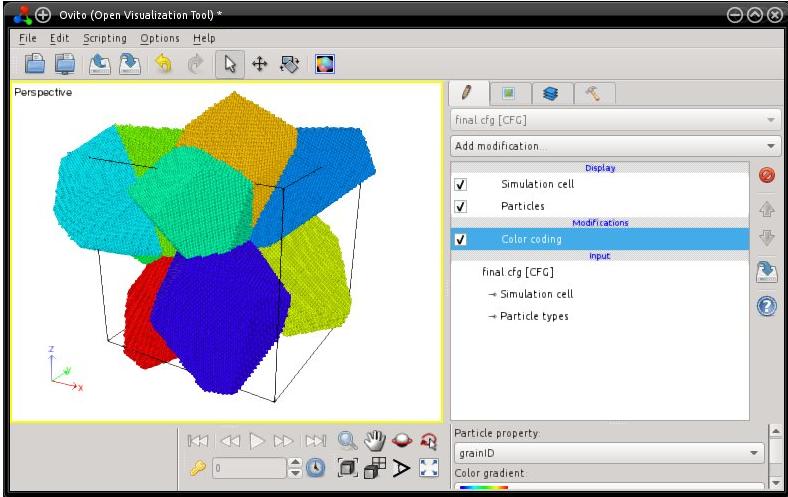
Notez que beaucoup de logiciels de visualisation (comme Atomeye) appliquent automatiquement des conditions aux limites périodiques, et remettent tous les atomes dans la boîte. Ce n'est pas le cas de Atomsk, aussi si vous ouvrez le fichier ("final.cfg") avec un éditeur de texte et parcourez les positions des atomes, vous trouverez beaucoup d'atomes qui sont à l'extérieur de la boîte (coordonnées négatives ou supérieures à 1). Sur ce sujet, il est recommandé de lire le tutoriel sur les conditions aux limites.
Si vous souhaitez appliquer des conditions aux limites périodiques, de sorte que dans le polycristal tous les atomes soient dans la boîte, alors vous devez le demander explicitement avec l'option "-wrap", par exemple :
atomsk --polycrystal aluminium.xsf polycrystal.txt final.cfg -wrap
Atomsk offre la possibilité de placer les nœuds en suivant un maillage pré-défini. Dans ce cas les positions des nœuds imiteront celles du réseau cristallin donné, et les orientations cristallographiques des grains seront aléatoires.
Pour utiliser cela, écrivez les dimensions de la boîte dans un fichier comme précédemment, puis écrivez le mot-clé "lattice" suivi du type de réseau cristallin : "bcc", "fcc", "diamond", ou "hcp". Par exemple, pour produire un polycristal dont les grains suivant un arrangement cubique centré (bcc) :
box 100 100 100
lattice bcc
Le polycristal peut ensuite être généré comme précédemment :
atomsk --polycrystal aluminium.xsf polycristal.txt bcc.cfg
Vous pouvez visualiser le résultat final "bcc.cfg" avec OVITO. Ci-dessous, les positions des atomes sont représentées telles que construites avec Atomsk à gauche, montrant qu'un grain est placé à l'origine et l'autre au centre de la boîte, imitant bien les sites d'un réseau cubique centré ; et sur la droite, les atomes ont été replacés dans la boîte en utilisant des conditions aux limites périodiques (dans OVITO utilisez "Wrap at periodic boundaries"), de montrant que les atomes remplissent bien la boîte.
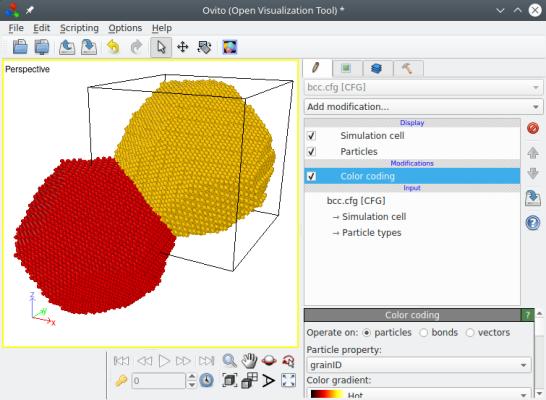
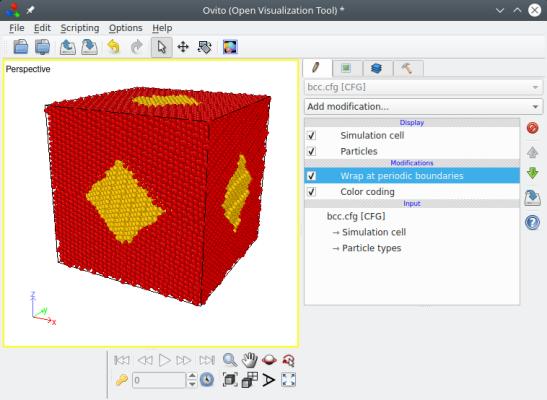
Certains points importants méritent d'être précisés ici. D'abord, les positions des nœuds imitent celles du réseau cubique centré (bcc), c'est-à-dire qu'un nœud est placé en (0,0,0), et le deuxième au milieu de la boîte. Si la boîte n'est pas cubique mais parallélépipédique, alors le deuxième nœud sera toujours placé au milieu de la boîte, produisant des grains de formes différentes. L'orientation des grains d'aluminium cependant, est toujours décidée aléatoirement par le programme. Si vous souhaitez mieux contrôler l'orientation des grains, lisez la section 5 ci-dessous.
Ensuite, le "réseau" utilisé pour le placement des nœuds, n'a rien à voir avec le réseau cristallin du matériau. Dans l'exemple précédent, le réseau cristallin de l'aluminium est bien cubique à faces centrées (cfc), seulement les nœuds du polycristal sont placés de sorte à imiter un réseau cubique centré (bcc). À la fin, chaque grain du polycristal contient bien un cristal d'aluminium cfc.
Vous pouvez essayer d'autres types de placements (fcc, diamond, hcp) et voir l'effet sur le polycristal final.
Parfois il est préférable de contrôler "manuellement" les positions et/ou les orientations cristallographiques des grains. Comme précédemment, le fichier texte doit commencer par les dimensions de la boîte. Puis, chaque grain doit être défini sur une ligne commençant par le mo-clé "node", suivi par la position du nœud (trois nombres réels) et son orientation cristalline. Par exemple :
box 200 180 210
node 0 0 0 [100] [010] [001]
node 40 80 60 56° -83° 45°
node 0.8*box 0.6*box 0.9*box [11-1] [112] [1-10]
node 50 5 60 [110] [1-10] [001]
node 0.75*box 0.3*box 0.45*box -31.4° 28.7° 90.0°
node 60 100 80 random
Un tel fichier de paramètres est interprété comme suit :
(1) La boîte finale aura pour dimensions 200 Å × 180 Å × 210 Å.
(2) Le premier grain sera placé à l'origine (0,0,0), et aura pour orientation cristallographique X=[100], Y=[010], Z=[001].
(3) Le deuxième grain sera positionné en (40;80;60) (en Å), et le cristal sera tourné de 56° autour de l'axe X, puis de -83° autour de Y, et enfin de 45° autour de Z.
(4) Le troisième grain sera positionné aux coordonnées réduites (0,8;0,6;0,9). Notez que le mot-clé "box" est utilisé pour spécifier des coordonnées réduites. Ce grain sera orienté X=[111], Y=[112], Z=[110].
(5) Le dernier grain sera positionné en (60;100;80) (en Å), et aura une orientation aléatoire.
Et ainsi de suite. Les diverses notations peuvent être mélangées dans le même fichier de paramètres, comme dans cet exemple, mais peut-être trouverez-vous plus pratique ou plus cohérent d'utiliser le même type de notation pour tous les grains, par exemple en donnant toutes les positions en Å (ou toutes en coordonnées réduites).
Après avoir généré votre fichier texte, exécutez Atomsk avec la même commande que précédemment :
atomsk --polycrystal aluminium.xsf polycristal.txt final.xsf cfg
Cette fois, Atomsk utilisera les positions et orientations que nous avons demandées, et le polycristal ressemblera à ceci :

Atomsk permet également de construire des polycristaux bidimensionnels, ou quasi-2D. Pour cel, il suffit de donner une valeur nulle à l'une des dimensions de la boîte, comme dans cet exemple :
box 200 200 0
random 12
Atomsk va automatiquement remplacer cette valeur nulle, par la taille de la "graine" dans la direction correspondante. Ici, notre graine est une maille élémentaire d'aluminium, de paramètre de maille 4.046 Å, et donc Atomsk ajustera automatiquement la taille de la boîte finale à 4.046 Å suivant la direction Z. Puisque le polycristal est bidimensionnel, tous les grains auront leur direction [001] alignée avec l'axe Z. Voici à quoi ressemble le système final :
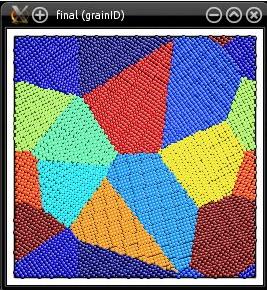
Comme précédemment, il est aussi possible d'utiliser le mot-clé "lattice" pour placer les nœuds suivant un motif pré-défini. Dans le cas d'un polycristal en 2-D, les nœuds sont placés suivant un réseau bidimensionnel.
Il est aussi possible de spécifier les positions et orientations des grains manuellement pour un polycristal 2-D, avec le mot-clé "node" comme expliqué dans la section 5 ci-dessus. Cependant, dans le cas d'un polycristal bidimensionnel, assurez-vous que toutes les coordonnées le long de l'axe court (ici Z) soient exactement les mêmes (par exemple mettez-les toutes à zéro), et n'entrez un angle de rotation qu'autour de l'axe court. Utiliser des positions différentes ou des rotations autour des autres axes brisera la symétrie du cristal le long de l'axe court, et risque de mener à des résultats incohérents.
Si vous souhaitez obtenir une autre direction cristalline suivant l'axe Z, alors la "graine" de départ doit avoir l'orientation cristallographique voulue. Dans notre exemple, nous devrions créer la maille de départ "aluminium.xsf" avec la bonne orientation cristallographique. Vous pouvez suivre ce tutoriel pour apprendre à générer des mailles orientées avec Atomsk.
La "graine" ne doit pas forcément être une maille élémentaire : en fait, n'importe quel système atomique peut être utilisé comme graine. Comme exemple, reprenons le système contenant une macle "Al_final.cfg", construit dans un tutoriel précédent. Utilisons-le pour construire un polycristal avec 12 grains (reprenez le fichier "polycristal.txt" de la section 2 ci-dessus) :
atomsk --polycrystal Al_final.cfg polycrystal.txt Al_twin_polyX.cfg
Au final, chaque grain du polycristal contiendra une macle :
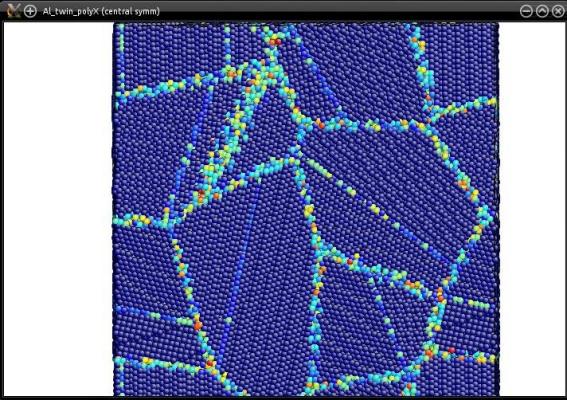
Plus généralement, n'importe quel système atomique peut être utilisé comme graine. Vous pouvez essayer avec un système contenant une dislocation (ou plusieurs), un défaut d'empilement, ou n'importe quel système que vous souhaitez.
La possibilité de construire des polycristaux bidimensionnels peut être exploitée pour construire des joints de grains, comme expliqué dans le tutoriel suivant.
Par ailleurs, il est aussi possible de générer des polycristaux multiphasés. Lisez ce tutoriel pour en savoir plus.