Atomsk
Le couteau suisse pour les simulations atomiques

Le couteau suisse pour les simulations atomiques




Dans ce tutoriel vous apprendre à construire des joints de grains avec le mode "--polycrystal".
▶ Pour plus d'information, référez-vous à la page de documentation correspondante.
En l'utilisant avec des paramètres spécifiques, le mode "--polycrystal" (décrit dans un précédent tutoriel) peut être utilisé pour générer des joints de grains particuliers. Après tout, un bi-cristal contenant un joint de grain peut être décrit comme un polycristal ne contenant que deux grains de position et orientation cristallographique particulières.
Les joints de flexion sont un type particulier de joints de grains, où les deux grains possèdent un axe cristallographique en commun, et sont inclinés l'un par rapport à l'autre autre par rotation autour de cet axe. Lorsque les deux grains sont tournés d'angles opposés, on dit que le joint de flexion est symétrique.
Les étapes pour construire un joint de flexion symétrique sont décrites par le schéma suivant (ici on considère l'axe cartésien Y normal au plan du joint, et l'axe Z normal à la figure) :
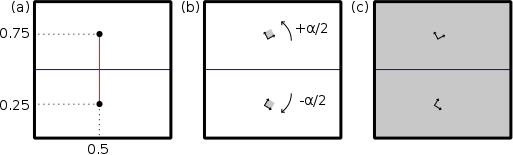
(a) Insérer deux nœuds aux coordonnées réduites (0,5;0,25;0) et (0,5;0,75;0). De cette façon, le joint de grains sera positionné au centre de la boîte finale (ligne bleue horizontale). Notez que, si des conditions aux limites périodiques sont utilisées suivant Y, alors il existera un autre joint en bord de boîte.
(b) Insérer des graines aux positions des nœuds. Tourner l'une d'elles d'un angle +α/2 et l'autre de −α/2 atour de l'axe Z, de sorte que la désorientation entre les deux grains soit α.
(c) Dupliquer la graine dans toutes les directions de l'espace, et couper chaque grain au niveau du joint.
Dans la suite, nous allons utiliser une maille élémentaire d'aluminium comme graine. Une telle maille peut être générée avec :
atomsk --create fcc 4.046 Al aluminium.xsf
À titre d'exemple, construisons un joint de flexion symétrique {210}[001] dans l'aluminium. Cela signifie que les deux grains on des surfaces {210} en commun, nous devons donc tourner les mailles d'un angle ±26.57° autour de l'axe Z. Créez un nouveau fichier texte, et écrivez-y les informations suivantes :
box 100 100 0
node 0.5*box 0.25*box 0 0° 0° -26.57°
node 0.5*box 0.75*box 0 0° 0° 26.57°
Ce fichier doit satisfaire au format du mode "--polycrystal", comme expliqué dans le tutoriel précédent. Un joint de grains est simplement un cas particulier d'un polycristal où seulement deux grains sont construits -chaque mot-clé "node" correspond à un grain. Le premier grain est placé en (0,5;0,25;0) et le deuxième en (0,5;0,75;0) comme expliqué précédemment. Le premier grain est tourné de 0° autour de l'axe X, 0° autour de Y, et −26.57° autour de Z ; de façon similaire, le deuxième grain est tourné de +26.57° autour de Z. Notez également que sur la première ligne, la dimension de la boîte suivant Z est nulle. Atomsk va automatiquement ajuster la taille de la boîte pour correspondre à la taille de la graine dans cette direction -dans notre cas, la boîte suivant Z deviendra aussi grande que la maille d'aluminium fournie au départ, autrement dit égale au paramètre de maille choisi 4.046 Å.
Maintenant, tout ce qu'il reste à faire est d'exécuter Atomsk en mode "--polycrystal", en utilisant le fichier "aluminium.xsf" comme graine, et les paramètres du fichier "polyX.txt" :
atomsk --polycrystal aluminium.xsf polyX.txt polycristal.cfg
Les instructions ci-dessus construisent bien deux grains avec l'orientation choisie, cependant la visualisation (par exemple avec Atomeye) montre qu'il y a des problèmes aux bords de la boîte. En effet, dans le fichier "polyX.txt" nous avons simplement défini une taille de boîte de 100 Å suivant X et Y, ce qui ne respecte pas la périodicité du réseau dans ces directions.
Suivant les axes cartésiens X et Y, le cristal a des directions de type <210>. Suivant ces axes, la période du cristal est égale à 4.046*√5, et donc les dimensions de la boîte doivent être un nombre entier de fois ce nombre pour respecter la périodicité. Par exemple :
box 9.047131 36.188524 0
node 0.5*box 0.25*box 0 0° 0° -26.57°
node 0.5*box 0.75*box 0 0° 0° 26.57°
Construire le polycristal avec ce fichier de paramètres au lieu du précédent, devrait donner un système compatible avec des conditions aux limites périodiques. Dans ce cas, les joints de grains en bord de boîte (suivant Y) sont équivalents au joint au centre de la boîte. Ici nous avons choisi des tailles différentes suivant X et Y, mais il est bien sûr possible d'utiliser les mêmes valeurs. Vous pouvez modifier ces valeurs, assurez-vous qu'elles soient un multiple entier de la période du système. Le fichier final "polycristal.cfg" peut être visualisé, par exemple avec Atomeye :

Notez que les dimensions de la boîte dépendent des directions cristallographiques, et donc de l'angle de désorientation, ils doivent donc être re-calculés pour chaque direction et désorientation choisies.
À titre d'exercices, vous pouvez essayer de construire des joints de grains de différentes orientations, par exemple {310}. Vous pouvez aussi construire des joints de flexion avec une direction différente suivant Z, par exemple en utilisant une maille d'aluminium orientée X=[110], Y=[001], Z=[110]. Enfin, vous pouvez aussi construire des joints de grains dans d'autres matériaux qui ont des réseaux cristallins différents, comme le fer cubique centré, le silicium, ou n'importe quel matériau, en changeant la graine, et en choisissant judicieusement les paramètres dans le fichier de paramètres ("polyX.txt").
Parfois, il est nécessaire de construire un système plus large contenant un joint de grains. Dans un tel cas, il est déconseillé d'écrire dans le fichier de paramètres ("polyX.txt") la taille de la grande boîte finale, car Atomsk peut avoir du mal à la remplir si elle doit contenir beaucoup d'atomes. Au lieu de cela, il est recommandé de construire un joint dans une boîte de taille minimale (comme nous l'avons fait ci-dessus), et ensuite, de la dupliquer pour parvenir à la taille du système final. Par exemple, nous pouvons dupliquer le joint de grains précédent 5 fois suivant X et 3 fois suivant Y&nbp;:
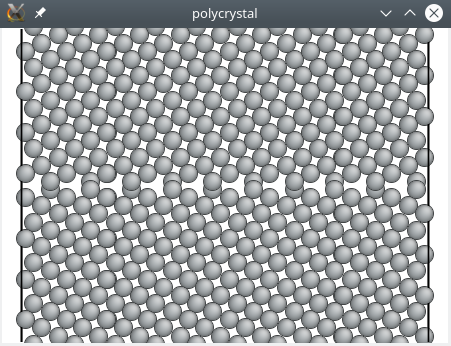
atomsk --polycrystal aluminium.xsf polyX.txt polycristal.cfg -dup 5 3 1
La visualisation nous montre que le système a bien été dupliqué :
Un autre type particulier de joints de grains, sont les joints de torsion, où les deux grains sont tournés l'un par rapport à l'autre autour de l'axe perpendiculaire au joint de grains. Dans ce cas, la taille de boîte suivant Y est un multiple entier du paramètre de maille, et les dimensions suivant X et Z dépendent de l'angle de désorientation.
À titre d'exemple, construisons un joint de torsion dans l'aluminium, en utilisant le même angle de rotation que précédemment. Nous savons que la taille de boîte suivant Y doit être un multiple du paramètre de maille, disons 10 fois pour faire simple, donc 40.46 Å. Suivant X et Z, la boîte doit avoir une taille multiple d'un vecteur [210] du réseau ; choisissons la plus petite taille, 9.047131 Å, dans chacune de ces directions. Enfin, les graines doivent être placées au milieu du plan XZ, c'est-à-dire aux coordonnées (½ ¼ ½) et (½ ¾ ½). Si nous incorporons tout cela dans un fichier de paramètre, il ressemblera à cela :
box 9.047131 40.46 9.047131
node 0.5*box 0.25*box 0.5*box 0° -26.57° 0°
node 0.5*box 0.75*box 0.5*box 0° 26.57° 0°
En exécutant la même commande que précédemment, nous obtenons cette fois un joint de torsion :
atomsk --polycrystal aluminium.xsf polyX.txt polycrystal.cfg
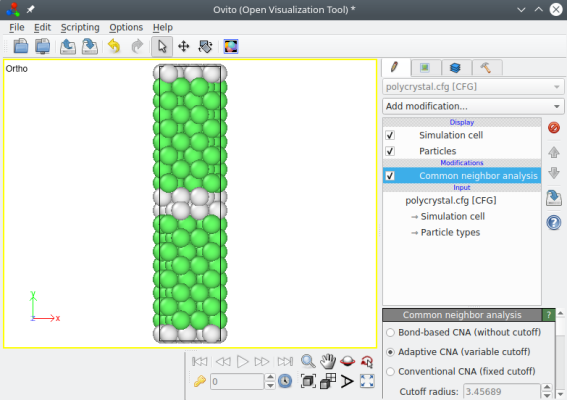
Ce système peut lui aussi être dupliqué après sa création, afin d'obtenir un système plus grand.