Atomsk
Le couteau suisse pour les simulations atomiques

Le couteau suisse pour les simulations atomiques




Ce tutoriel explique comment Atomsk gère la boîte, les positions des atomes, et les conditions aux limites.
Pour n'importe quel système atomique, Atomsk travaille avec au moins deux types de données :
Certains systèmes peuvent contenir davantage d'informations (comme les vitesses ou les forces sur les atomes), mais un système atomique doit forcément contenir au moins les vecteurs de boîtes et positions atomiques. Par défaut, Atomsk travaille avec les coordonnées cartésiennes des atomes, qui sont absolues et indépendantes des vecteurs de boîte.
Atomsk traite les vecteurs de boîte comme des vecteurs à part entière, c'est-à-dire que chaque vecteur de boîte peut avoir des composantes suivant les axes cartésiens X, Y et Z, et ils peuvent former n'importe quel angle entre eux (ils ne doivent pas forcément être orthogonaux). Dit autrement, dans le cas le plus général les vecteurs de boîte sont H1=(x1 y1 z1), H2=(x2 y2 z2), H3=(x3 y3 z3).
Cela vaut le coup d'être mentionné, car ce n'est pas forcément le cas de tous les codes de simulation ou de visualisation. Par exemple, LAMMPS requiert que le premier vecteur de boîte ait des coordonnées y1=z1=0, et que le second vecteur de boîte ait z2=0. Cela veut dire qu'il est impossible d'écrire certaines composantes des vecteurs de boîte dans un fichier au format LAMMPS, et qu'il peut être nécessaire d'appliquer une rotation au système pour satisfaire les exigences de LAMMPS. Pour en savoir plus vous pouvez lire ce tutoriel.
Un autre exemple est le format natif de VESTA, où les vecteurs de boîte sont enregistrés en notation "boîte conventionnelle" : seules les normes (longueurs) des vecteurs de boîte a, b, c, et les angles qu'ils forment entre eux α, β, γ, peuvent être écrits dans un fichier *.vesta. Ainsi, lors de la visualisation d'un système, VESTA suppose toujours que le premier vecteur est aligné avec l'axe cartésien X, et que le second vecteur est dans le plan XY. Donc, en écrivant des fichiers dans ce format, toute information suite à une rotation rigide du système est tout simplement perdue.
Au sein de Atomsk, l'origine de la boîte est toujours confondue avec l'origine des coordonnées cartésiennes (0 0 0). Encore une fois, ce n'est pas forcément le cas dans tous les codes. Certains codes autorisent une origine différente (par exemple dans LAMMPS elle prend les coordonnées xmin, ymin, zmin). Quand Atomsk lit un tel fichier, il translate automatiquement tout le système (boîte et positions atomiques) pour que l'origine de la boîte soit (0 0 0).
Atomsk travaille avec les coordonnées cartésiennes des atomes (habituellement en Å), et traite les atomes séparément de la boîte. Atomsk n'applique pas automatiquement des conditions aux limites périodiques les atomes peuvent avoir n'importe quelles coordonnées (x y z), même si elles sont situées en dehors de la boîte.
Pour l'exemple, créons une maille élémentaire d'aluminium :
atomsk --create fcc 4.046 Al aluminium.xsf
Si vous ouvrez le fichier "aluminium.xsf" avec un éditeur de texte, vous verrez qu'il contient trois vecteurs de boîte, et les coordonnées cartésiennes des atomes (en Å) :
# Fcc Al oriented X=[100], Y=[010], Z=[001].
CRYSTAL
PRIMVEC
4.04600000 0.00000000 0.00000000
0.00000000 4.04600000 0.00000000
0.00000000 0.00000000 4.04600000
CONVVEC
4.04600000 0.00000000 0.00000000
0.00000000 4.04600000 0.00000000
0.00000000 0.00000000 4.04600000
PRIMCOORD
4 1
13 0.00000000 0.00000000 0.00000000
13 2.02300000 2.02300000 0.00000000
13 0.00000000 2.02300000 2.02300000
13 2.02300000 0.00000000 2.02300000
Dans ce fichier, tous les atomes sont à l'intérieur de la boîte. Maintenant, que se passe-t-il si on applique un large déplacement aux atomes pour les faire sortir de la boîte ? Essayons avec l'option "-shift" :
atomsk aluminium.xsf -shift 10 10 10 Al_shift.xsf
Le nouveau fichier "Al_shift.xsf" ressemble à cela :
# Fcc Al oriented X=[100], Y=[010], Z=[001].
CRYSTAL
PRIMVEC
4.04600000 0.00000000 0.00000000
0.00000000 4.04600000 0.00000000
0.00000000 0.00000000 4.04600000
CONVVEC
4.04600000 0.00000000 0.00000000
0.00000000 4.04600000 0.00000000
0.00000000 0.00000000 4.04600000
PRIMCOORD
4 1
13 10.00000000 10.00000000 10.00000000
13 12.02300000 12.02300000 10.00000000
13 10.00000000 12.02300000 12.02300000
13 12.02300000 10.00000000 12.02300000
Comme demandé, toutes les coordonnées ont été modifiées de 10 Å.
Ce comportement est important Atomsk conserve les coordonnées cartésiennes des atomes. Il n'applique jamais de conditions aux limites périodiques de façon automatique, ni ne replace les atomes à l'intérieur de la boîte. Gardez cela en tête lorsque vous construisez vos systèmes quand les atomes sont déplacés et sortent de la boîte, ne vous attendez pas à ce que Atomsk applique automatiquement des conditions aux limites périodiques !
Si vous souhaitez appliquer des conditions aux limites périodiques, alors vous devez le demander explicitement, avec l'option "-wrap" comme expliqué ci-dessous.
La visualisation de systèmes où certains atomes sont en dehors de la boîte peut être trompeuse. Convertissons le système précédent (celui qui a été translaté) au format CFG pour le visualiser :
atomsk Al_shift.xsf cfg
Ceci va produire un fichier nommé "Al_shift.cfg". Visualisons-le avec OVITO, comme le montre l'exemple ci-dessous :
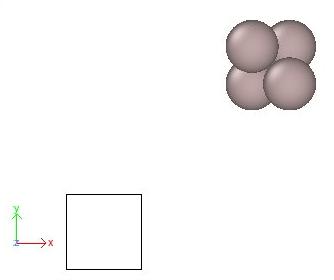
OVITO affiche bien le repère cartésien (flèches colorées labelisées x, y, z), la boîte de simulation (rectangle noir), et les atomes (sphères grises). Effectivement, les atomes sont en dehors de la boîte, comme on s'y attendait avec les commandes exécutées précédemment.
Maintenant, visualisons exactement le même fichier ("Al_shift.cfg") avec Atomeye :
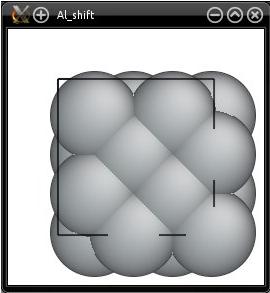
Cette fois, deux choses ont l'air incorrectes. Premièrement, Atomeye affiche les atomes comme s'ils étaient dans la boîte. Pourquoi ? Parce que Atomeye applique automatiquement des conditions aux limites périodiques, et replace les atomes dans la boîte. La deuxième chose est que notre système ne contient que quatre atomes, mais Atomeye en affiche bien plus que cela. C'est parce que Atomeye considère que notre système est "trop petit", alors il le duplique automatiquement deux fois suivant chaque direction, comme expliqué dans le tutoriel précédent.
En résumé, il est important que vous connaissiez les limitations et les automatismes des logiciels de visualisation que vous employez. Certains ne montrent pas les véritables coordonnées des atomes telles qu'elles sont écrites dans le fichier. Au lieu de cela, ils appliquent automatiquement (et sans vous prévenir) des conditions aux limites périodiques. Parfois c'est pratique pour faciliter la visualisation, mais parfois cela peut aussi être trompeur et fausser votre interprétation des données. Soyez donc très prudent lorsque vous visualisez vos systèmes atomiques. Lisez la documentation pour savoir quelles conditions aux limites ils appliquent, et pour savoir s'ils font d'autres opérations silencieusement.
Nous l'avons dit, Atomsk n'applique pas automatiquement de conditions aux limites périodiques, ni ne replace les atomes dans la boîte. Si vous souhaitez que tous les atomes soient remis dans la boîte, alors vous devez le demander explicitement avec l'option "-wrap". Par exemple, demandons à Atomsk de lire le fichier précédent "Al_shifted.cfg", où tous les atomes sont hors de la boîte, et remettons-les dans la boite :
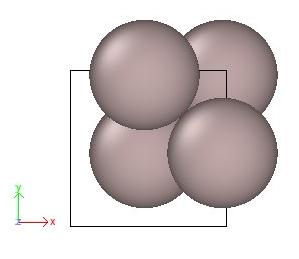
atomsk Al_shift.cfg -wrap Al_wrap.cfg
L'option "-wrap" applique à chaque atome une translation d'un ou plusieurs vecteurs de boîte, jusqu'à ce que toutes leurs coordonnées tombent à l'intérieur de la boîte. La visualisation avec OVITO confirme que les atomes sont désormais à l'intérieur de la boîte :
Il est possible de demander à Atomsk de convertir toutes les coordonnées des atomes de cartésien (par défaut) à réduites (ou fractionnaires), grâce à l'option "-fractional" :
atomsk Al_shift.xsf -fractional Al_frac.xyz
Les coordonnées réduites, ou fractionnaires, sont exprimées en fonction des vecteurs de boîte. Si les atomes sont dans la boîte, alors ces coordonnées prennent des valeurs entre 0 et 1. Cependant, si un atome est en dehors de la boîte, alors ses coordonnées réduites peuvent être négatives, ou plus grandes que 1.
Rappelez-vous que l'option "-fractional" ne replace pas les atomes dans la boîte, elle ne fait que diviser leurs coordonnées par les vecteurs de boîte.
Comme expliqué au début, Atomsk requiert toujours au moins les vecteurs de boîte, ainsi que les positions atomiques et les numéros atomiques. C'est le strict minimum nécessaire. Ensuite, d'autres informations peuvent exister.
Parfois, le fichier d'entrée contient des données supplémentaires sur les atomes. Par exemple, les fichiers de sortie de LAMMPS peuvent contenir le "type" des atomes, ou la force s'appliquant sur chaque atome, ou son énergie potentielle, et ainsi de suite. Dans ce cas, Atomsk enregistre toutes ces données afin qu'elles puissent être manipulées, et éventuellement transférées au fichier de sortie. Cependant, notez que certains fichiers de sortie n'acceptent pas les données additionnelles, auquel cas Atomsk ne peut pas les écrire, et ces données sont alors perdues.
Dans certaines simulations, un système peut aussi contenir des coquilles ioniques (dans le cadre d'un modèle ionique cœur-coquille). Atomsk enregistre aussi les positions des coquilles, et la plupart des options (comme "-duplicate", "-shift", etc.) sont compatibles avec les coquilles. Vous pouvez lire ce tutoriel pour savoir comment Atomsk traite les cœurs et coquilles ioniques.
Quand vous construisez un système avec Atomsk, vous pouvez déplacer les atomes, insérer une dislocation, ou appliquer d'autres transformations qui peuvent déplacer des atomes en-dehors de la boîte. Gardez en tête que Atomsk n'applique pas automatiquement de conditions aux limites périodiques, et donc, selon les transformations (options) que vous appliquez, certains atomes peuvent se trouver en-dehors de la boîte.
S vous voulez être sûr que tous les atomes sont à l'intérieur de la boîte, utilisez simplement l'option "-wrap".