Atomsk
Le couteau suisse pour les simulations atomiques

Le couteau suisse pour les simulations atomiques




Ce tutoriel donne des exemples pour appliquer une translation ou une fonction de déplacement aux atomes.
▶ Pour plus d'informations, lisez la documentation des options "-shift", "-rotate", "-select", et "-properties".
Pour ce tutoriel, nous allons utiliser une super-cellule d'aluminium :
atomsk --create fcc 4.046 Al -duplicate 20 20 1 Al_supercell.cfg
L'option "-shift" est conçue pour translater facilement des atomes. Elle requiert seulement les trois composantes cartésiennes du vecteur de translation, en angströms :
atomsk Al_supercell.cfg -shift 10.5 20.6 0.0 shift.cfg
Avec cette commande, tous les atomes sont translatés comme on peut le voir avec OVITO :
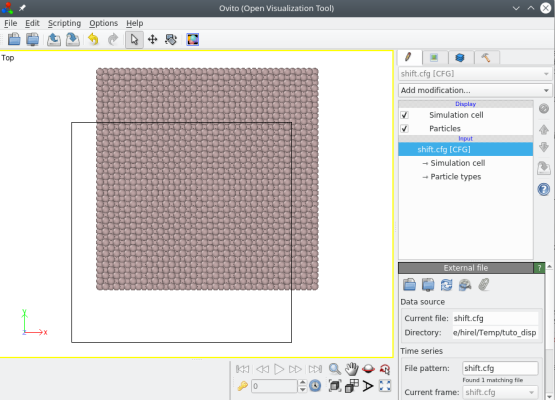
ⓘ Notez que la boîte n'est pas translatée, parce que dans Atomsk l'origine de laboîte est toujours confondue avec l'origine des coordonnées cartésiennes (0,0,0).
Cette option peut aussi être utilisée pour translater uniquement les atomes situés au-dessus ou en-dessous d'un plan donné. Pour cel, ajoutez le mot-clé "above" (au-dessus) ou "below" (en-dessous), la distance et la normale au plan, suivi du vecteur de translation, par exemple :
atomsk Al_supercell.cfg -shift above 40 Y 10.5 0 0 shift.cfg
Seuls les atomes dont la coordonnée Y est supérieure à 40 Å sont déplacés :
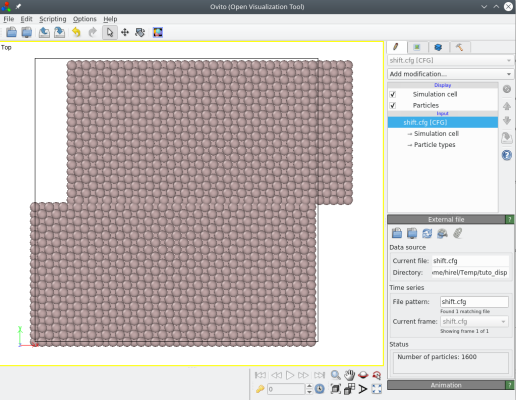
Il est aussi possible de donner la distance du plan de coupure avec une opération (par exemple "0.5*box"), ou de donner la normale au plan comme une direction cristalline, comme [110] ou [-111]. Veuillez vous référer à la page de documentation de l'option "-shift" pour plus d'informations.
Enfin, si l'option "-select" est utilisée pour sélectionner une partie du système, alors seuls les atomes sélectionnés sont translatés. Par exemple, sélectionnons les atomes à l'intérieur d'une boîte et translatons-les :
atomsk Al_supercell.cfg -select in box 30 30 -10 100 60 10 -shift 50 0 0 shift.cfg
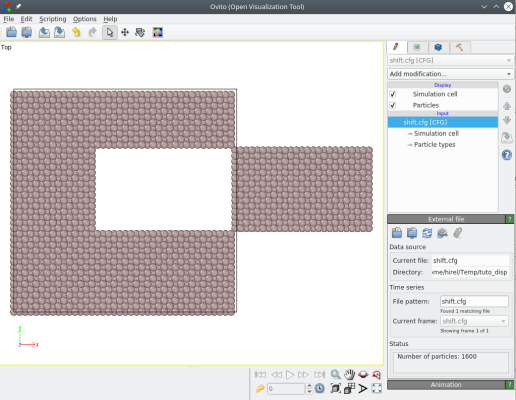
Vous pouvez aussi utiliser l'option "-select" pour sélectionner un ou plusieurs atomes en donnant leurs indices. Par exemple, si vous souhaitez translater les atomes numéros 45 et 86, vous pouvez utiliser :
atomsk Al_supercell.cfg -select 45,86 -shift 50 0 0 shift.cfg
Vous pouvez parcourir la documentation de l'option "-select" pour en découvrir les différentes possibilités.
L'option "-rotate" permet d'appliquer une rotation du système autour d'un axe donné :
atomsk Al_supercell.cfg -rotate Z 15 rotated.cfg
La commande ci-dessus tourne l'ensemble du système autour de l'axe Z de 15 degrés :
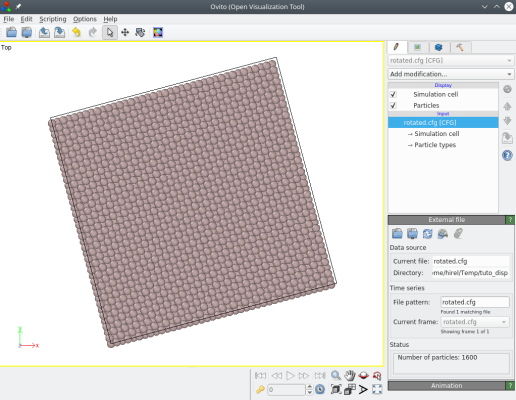
ⓘ Notez que les atomes et la boîte ont tourné, de sorte que les vecteurs de boîte ne sont plus alignés avec les directions cartésiennes X et Y.
Si vous souhaitez tourner un groupe d'atomes autour de son centre de masse, alors sélectionnez-les avec l'option "-select", puis utilisez l'option "-rotate" avec le mot-clé "com". Par exemple, appliquons une rotation à une sphère d'atomes au milieu de la boîte :
atomsk Al_supercell.cfg -select in sphere 0.5*box 0.5*box 0.5*box 20 -rotate com Z 33 shift.cfg
Notez que quand un groupe d'atomes est sélectionné, alors la boîte reste identique (elle ne subit pas la rotation), seuls les atomes sélectionnés sont tournés.
ⓘ Il est possible de spécifier la position du centre de la sphère en angströms, ou bien en coordonnées réduites avec le mot-clé "box" comme dans l'exemple ci-dessus. Cependant le rayon de la sphère doit être donné en angströms.
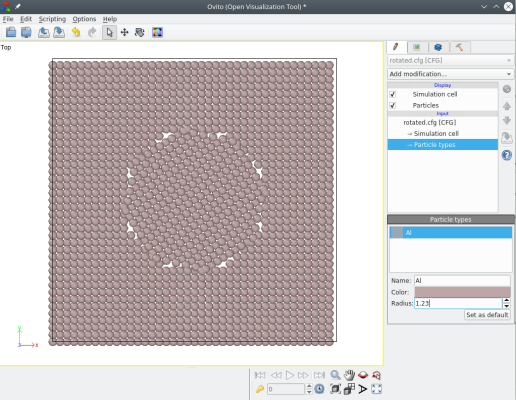
En plus des trois axes cartésiens (X, Y, ou Z), l'axe de rotation peut aussi être une direction cristallographique. Veuillez lire la page de documentation de l'option "-rotate".
L'option "-disturb" applique des déplacements aléatoires aux atomes. Ces déplacements suivent une distribution normale, et sont ajustés de sorte qu'il n'y ait pas de translation globale du système. Les normes des déplacements sont comprises entre zéro (aucun déplacement) et la valeur maximale que vous fournissez.
Par exemple, appliquons des déplacements aléatoires avec un maximum de 1.5 Å :
atomsk Al_supercell.cfg -disturb 1.5 random.cfg
La visualisation avec OVITO montre que Atomsk a appliqué des déplacements aléatoires (ux,uy,uz) :
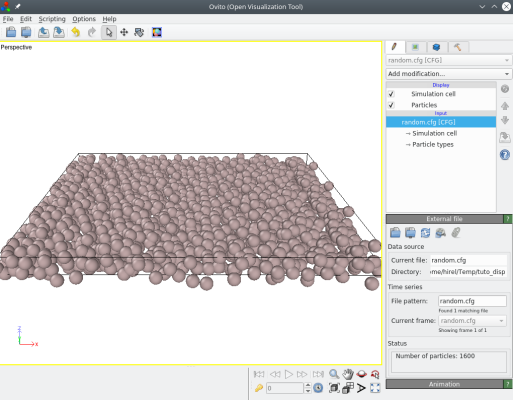
Il est aussi possible de fournir trois valeurs, correspondant aux déplacements maximaux suivant chaque direction cartésienne. Par exemple, pour appliquer des déplacements aléatoires uniquement suivant Z, nous pouvons utiliser :
atomsk Al_supercell.cfg -disturb 0 0 1.0 random2.cfg
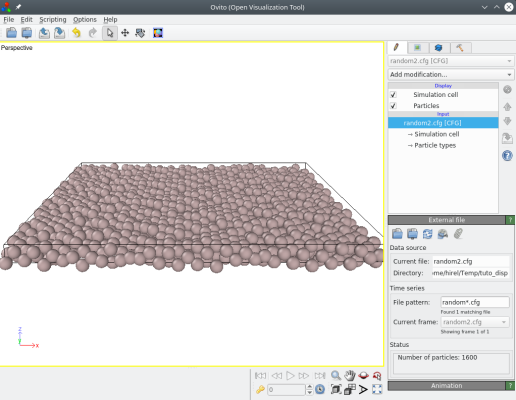
Là encore, vous pouvez essayer différentes valeurs, et visualiser les résultats.
Si vous souhaitez appliquer un déplacement spécifique et différent pour chaque atome, alors vous pouvez utiliser l'option "-properties". Cette option lit un fichier contenant des informations sur les atomes. Ici, nous allons l'utiliser pour lire les déplacements que nous voulons appliquer aux atomes.
D'abord, écrivons les déplacements voulus dans un fichier texte. Ce fichier doit contenir le mot-clé "displacement", suivi de lignes indiquant l'indice de l'atome, et le déplacement dx dy dz à appliquer. Voici un exemple (les lignes commençant par le symbole # sont des commentaires et sont ignorées par Atomsk) :
# Déplacements atomiques (en angströms)
displacement
1 0.2 0.2 2.5
2 -1.2 0.1 -2
3 -0.1 0.08 -1.3
30 -0.6 0.08 3.9
Il n'est pas obligatoire d'écrire les déplacements pour tous les atomes, seulement des atomes que vous voulez déplacer. De même, les indices des atomes peuvent être donnés dans n'importe quel ordre (ils n'ont pas forcément besoin d'être dans un ordre croissant). Le fichier peut contenir les déplacements seulement pour certains atomes, et dans n'importe quel ordre -Atomsk va immédiatement appliquer chaque déplacement à l'atome correspondant. Vous pouvez essayer d'ajouter d'autres lignes au fichier ci-dessus, pour changer les déplacements ou déplacer d'autres atomes.
Puis, utilisez l'option "-properties" pour lire ces déplacements et les appliquer au système atomique :
atomsk Al_supercell.cfg -properties disp.txt disp.cfg
Le résultat peut être visualisé avec OVITO, où on voit que les atomes demandés ont effectivement été déplacés par Atomsk :
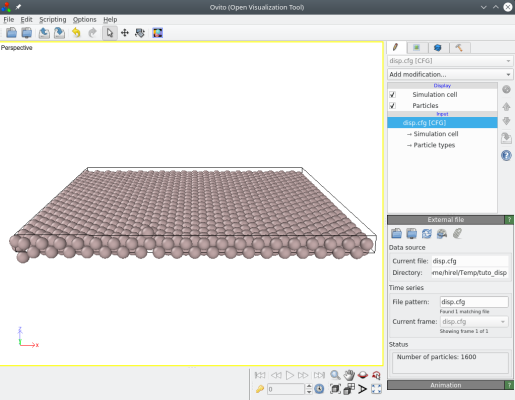
L'option "-properties" permet aussi de déplacer les atomes suivant une fonction mathématique. Pour y parvenir, utilisez le mot-clé "displacement function" dans le fichier texte, puis donnez les fonctions donnant les déplacements ux, uy et/ou uz. Chaque fonction doit commencer par "ux=" (ou "uy=" ou "uz="), suivi d'une fonction qui peut contenir des nombres, des opérateurs (+, -, *, /, ^), des fonctions mathématiques (cos, sin, tan, exp, sqrt...), ou la constante "pi". Référez-vous à la documentation pour une description complète.
À titre d'exemple, écrivons une fonction qui applique des déplacements uz (c'est-à-dire uniquement suivant la direction cartésienne Z) :
# Fonctions de déplacements pour les atomes
displacement function
uz = 10*cos(4*pi*x/Hx)
Le mot-clé "pi" est automatiquement replacé par la valeur de π, Hx par la longueur du premier vecteur de boîte, et x par la coordonnée de chaque atome. Comme précédemment, invoquons l'option "-properties" pour appliquer ces déplacements :
atomsk Al_supercell.cfg -properties disp.txt fonction.cfg
Le fichier résultant "fonction.cfg" peut être visualisé avec OVITO. Effectivement, Atomsk a bien déplacé les atomes en suivant la fonction donnée :
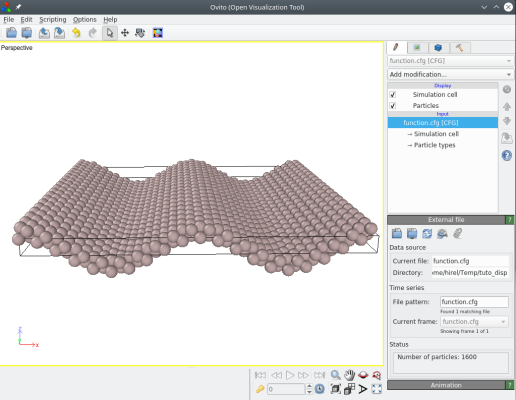
Il est bien sûr possible de modifier la fonction, de sorte que les déplacements uz dépendent aussi de la coordonnée y des atomes, par exemple :
# Fonctions de déplacements pour les atomes
displacement function
uz = 2*cos(4*pi*x/Hx) + 2*cos(4*pi*y/Hy)
Exécutons la même commande que précédemment :
atomsk Al_supercell.cfg -properties disp.txt fonction2.cfg
Puis ouvrons le nouveau fichier "fonction2.cfg" avec OVITO (ici les atomes sont colorés suivant leur coordonnée Z pour rendre les déplacement plus visibles) :
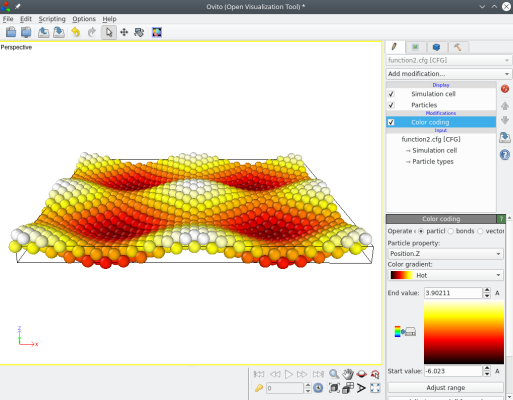
Vous pouvez modifier la fonction comme vous le souhaitez, par exemple pour changer sa périodicité remplacez "4*pi" par "6*pi" ou "2*pi", et visualisez le résultat. Vous pouvez aussi ajouter des fonctions de déplacement ux et uy. Tout ce que vous avez à faire, c'est modifier le fichier texte, et ensuite exécutez Atomsk pour appliquer les déplacements.
ⓘ Pour connaître la liste des fonctions mathématiques supportées, veuillez lire la documentation de l'option "-properties".
Les limites de cette option méritent d'être mentionnées. Cette option peut être lente, car Atomsk doit évaluer la fonction indépendamment pour chaque atome. Cette option est parallélisée, ce qui veut dire qu'elle utilisera tous les cœurs du processeur de votre machine pour plus d'efficacité. Malgré cela, si vous écrivez une fonction très compliquée (avec de nombreux opérateurs, fonctions imbriquées, parenthèses, etc.), alors Atomsk peut mettre beaucoup de temps à la calculer. Dans certains cas, il est même possible que Atomsk ne parvienne pas à interpréter certaines fonctions.
Comme on peut le voir avec les exemples ci-dessus, après avoir appliqué un déplacement, certains atomes peuvent se retrouver en-dehors de la boîte. Notez que Atomsk n'applique pas de conditions aux limites périodiques automatiquement. Si vous voulez les appliquer, alors vous devez le demander explicitement avec l'option "-wrap". Toutefois, selon les déplacements qui ont été appliqués, remettre les atomes dans la boîte peut provoquer le chavauchement de certains atomes.
Dans ce tutoriel nous avons utilisé une super-cellule "plate", c'est-à-dire quasiment à 2 dimensions, pour faciliter la visualisation des déplacements. Bien entendu, tous les concepts décrits ci-dessus se généralisent à des systèmes différents, par exemple au graphène, ou à des systèmes tridimensionnels -la seule limite est votre imagination.