Atomsk
The Swiss-army knife of atomic simulations

The Swiss-army knife of atomic simulations




This tutorial explains how to define, add or modify auxiliary properties with Atomsk.
▶ For more information, refer to the corresponding documentation page.
The minimum information to define an atomic system consists of atom species and their positions. However it is often useful to define additional information. In Atomsk, this can be achieved thanks to the option "-properties", which reads properties from a text file.
Atomsk distinguishes between two types of properties: those that take a different value for each atom, referred to as "per-atom properties" or "auxiliary properties"; and properties that apply to the whole system.
An important thing to understand, is that all file formats do not implement auxiliary properties.
So, if you wish to keep auxiliary properties when converting files, make sure that the target output format supports the properties that you defined for your system. Otherwise, if the output format offers no support for auxiliary properties, then Atomsk cannot write them and they will simply be lost. In order to know what auxiliary properties are supported by which format, please refer to the list of formats supported by Atomsk.
When reading an input file, Atomsk tries to load auxiliary properties as well. There again, refer to the list of formats to know what data is loaded from each file format. When converting from one format into another, Atomsk tries to pass along the auxiliary properties whenever possible. However, if the target output format does not support the same auxiliary properties as the input format, then they cannot be transferred and will simply be lost.
In the following, the CFG format will be largely used, because it allows to store any property, and can easily be visualized with Atomeye or OVITO.
Let us start with a property that is defined for each atom. As an example, assume that we want to construct an ionic crystal, for which the electric charge of each atom must be defined. Let us create a crystal of rocksalt NaCl, just like in a previous tutorial:
atomsk --create rocksalt 5.64 Na Cl cfg
Now if you open the resulting file "NaCl.cfg" with a text editor, you will see that the atoms do not have an electric charge. Indeed, by default Atomsk only generates the atom positions, and has no idea if the crystal is ionic or not, or what the charges should be. Hence it is up to the user to set up the ionic charges.
To achieve that, let us write the charges corresponding to each chemical species in a text file. The text file must contain the keyword "charge", and then on each line, a chemical species and the corresponding charge (in units of the elementary charge e):
# Charges for NaCl crystal
charge
Na 1.0
Cl -1.0
Then when creating the NaCl crystal, use the option "-properties" to read the charges:
atomsk --create rocksalt 5.64 Na Cl -properties charge.txt cfg
Now if you open the file "NaCl.cfg" with a text editor, you can notice the line "auxiliary[0] = q", and each line contains four numbers: the position x, y, z of the atom, and its electric charge. You may also open this file with OVITO and colour atoms according to the property "Charge":
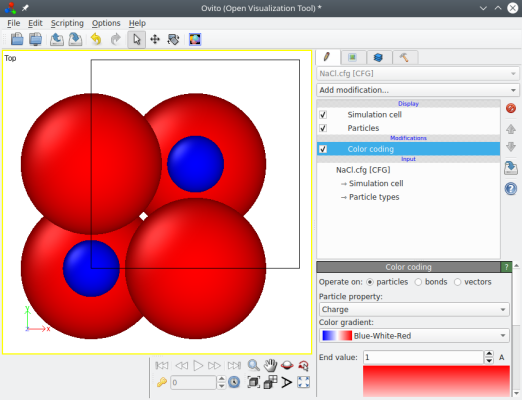
If you run the same command as above, replacing "cfg" with "lammps" as an output, then Atomsk will produce a LAMMPS data file with the atom style "charge" (instead of "atom" by default). You may read more about this in the tutorial for LAMMPS.
The user can define any per-atom property, thanks to the keyword "auxiliary" and the name of the auxiliary property. Then, each line must contain an atom index (integer number) and the corresponding value of the property. The index of atoms may appear in any order. If an atom index does not appear, the property automatically takes the value 0 for that atom.
For the sale of the example, assume that the atoms energy was computed (by a molecular dynamics code or any other script). As stated before, the text file must contain the keyword "auxiliary" and the name of the property (here we just name it "my_energy"), and in the following lines, an atom index and the value of the property:
auxiliary my_energy
2 -20.3
3 -20.4
4 -19.9
6 -19.86
15 -20.4
9 -19.9
atomsk --create rocksalt 5.64 Na Cl -duplicate 2 2 2 -properties energies.txt cfg
You may open the final file "NaCl.cfg" with a text editor and verify that the energies are there. This file may also be visualized with OVITO as shown below: add "Colour coding" and select the property "my_energy".
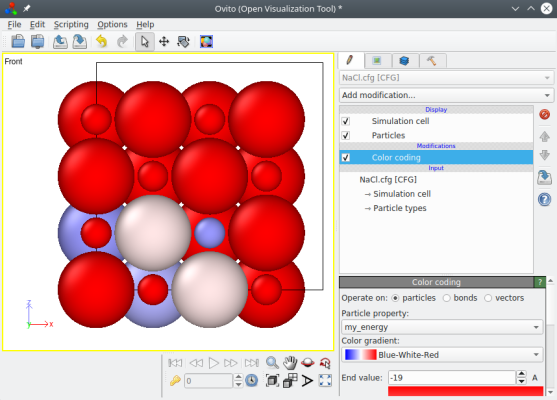
The option "-properties" is quite versatile, and allows you to add any custom property to an existing system.
It is also possible to define properties that do not apply to each atom individually, but rather to the system as a whole. This is the case, for example, if you wish to modify the dimensions of the box, without changing the positions of atoms. In this case, write the keyword "supercell" followed by the components of the three vectors of the box in a text file:
# New box vectors
supercell
5.64 0.00 0.00
0.00 5.64 0.00
0.00 0.00 5.70
And then use the option "-properties" to apply it:
atomsk NaCl.cfg -properties newbox.txt tetragonal.cfg
Note that only the box vectors are changed, while the atoms keep the same Cartesian positions.
Another system-wide property is the elastic tensor. To learn more about it, you may refer to the documentation, and follow the tutorial about dislocations in anisotropic medium.
After running a simulation or analysis, files can contain a lot of auxiliary properties, some of which are unnecessary. Atomsk can remove some properties with the option "-remove-property" followed by the property name. For example, if you wish to get rid of electric charges you can use:
atomsk NaCl.cfg -remove-property q final.cfg
If you wish to remove all auxiliary properties from the system:
atomsk NaCl.cfg -remove-property all final.cfg