Atomsk
Le couteau suisse pour les simulations atomiques

Le couteau suisse pour les simulations atomiques




Ce tutoriel explique comment construire une dislocation vis ½[110] dans l'aluminium. Il est recommandé d'être familier avec la théorie des dislocations pour suivre ce tutoriel.
▶ Pour plus d'information, référez-vous à la page de documentation correspondante.
L'aluminium est un métal cubique à faces centrées (cfc), dans lequel les systèmes de glissement sont de type ½⟨110⟩{111}. Supposons que nous voulions construire un système ne contenant qu'une seule dislocation vis, alignée avec l'axe cartésien Z, de vecteur de Burgers b=½[110], dans un plan {111} normal à Y, comme le montre le schéma ci-dessous :
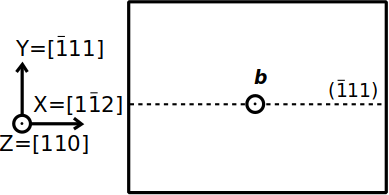
Avec ces informations, nous pouvons définir l'orientation du cristal dont nous avons besoin. Le vecteur de Burgers est parallèle à la ligne de dislocation, donc la direction [110] du cristal doit être suivant Z. Le plan de glissement doit être normal à Y, donc nous devons avoir Y=[111]. La dernière direction doit donc être X=[112]. Construisons cette super-cellule, en utilisant comme paramètre de maille a=4.046 Å :
atomsk --create fcc 4.046 Al orient [1-12] [-111] [110] -duplicate 40 20 1 Al_supercell.xsf
Le vecteur de Burgers ½[110] a pour norme |b|=a/√2, ce qui vaut environ 2.860954 Å avec le paramètre de maille que nous avons choisi. Bien entendu, si vous travaillez avec un paramètre de maille différent, alors vous devrez calculer la norme du vecteur de Burgers correspondant.
Nous introduisons la dislocation vis avec Atoms, en utilisant l'option "-dislocation", suivie de la position de la dislocation dans le plan XY (ici nous la plaçons au centre de la boîte), le mot-clé "screw", la direction de la ligne (Z), la normale au plan de glissement (Y), et la norme du vecteur de Burgers :
atomsk Al_supercell.xsf -dislocation 0.51*box 0.501*box screw Z Y 2.860954 Al_screw.xsf cfg
ⓘ La valeur du vecteur de Burgers doit être donné précisément, et doit être cohérent avec le paramètre de maille que vous avez choisi. Atomsk ne va pas ajuster "auto-magiquement" sa valeur.
ⓘ La position de la dislocation peut être entrée comme une fraction des vecteurs de boîte comme ci-dessus ("0.51*box"), ou bien directement en Angströms. La position donnée ne devrait pas correspondre exactement à la position d'un atome, sinon cet atome aurait un déplacement infini. Si vous obtenez des résultats étranges comme des discontinuités, des trous ou de larges déplacements, essayez de changer légèrement la position de la dislocation.
Atomsk calcule aussi les composantes du tenseur des contraintes théoriques (d'après la théorie de l'élasticité), qui peuvent ensuite être visualisées avec Atomeye ou OVITO. Par exemple, l'image ci-dessous montre les composantes σxz et σyz des contraintes :
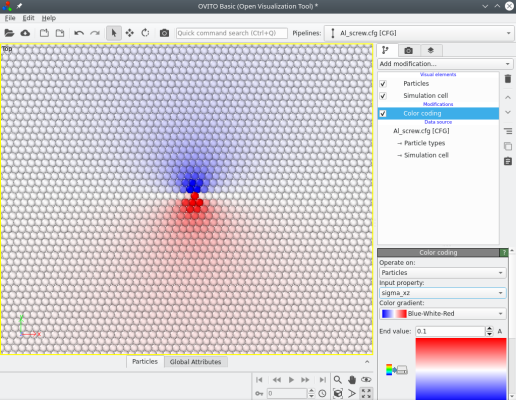
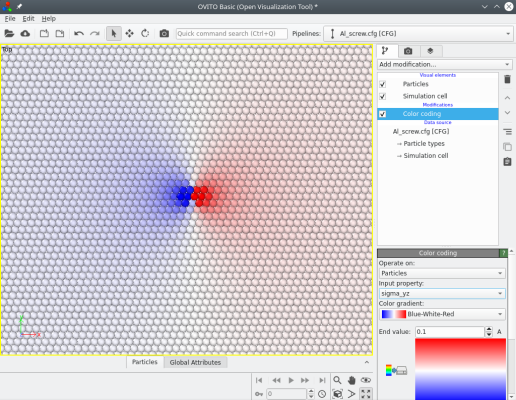
Lorsqu'une dislocation vis est introduite, le nombre d'atomes reste constant. Le système reste périodique le long de la lgne de dislocation (ici suivant Z), mais les déplacements dus à la dislocation brisent la périodicité suivant les autres directions (X et Y). Cela peut être corrigé de différentes façons :
ⓘ Atomsk n'applique aucune de ces solutions particulières automatiquement, c'est donc à l'utilisateur (vous) de décider quelle solution correspond le mieux à vos besoins, et de la mettre en œuvre.
Il est possible de restaurer la périodicité du cristal le long de la direction de glissement (ici la direction X). En effet, aux bords de la boîte, les atomes ont été déplacés suivant Z de +b/2 au-dessus du plan de glissement, et -b/2 en-dessous. Ainsi, ajouter une composante suivant Z égale à b/2 au premier vecteur de la boîte permet de recouvrer la périodicité suivant X. Cela peut être fait avec l'option "-cell", qui permet de modifier les composantes des vecteurs de boîte :
atomsk Al_screw.xsf -cell add 1.430477 zx Al_screw_cell.cfg
De façon alternative, vous pouvez aussi changer la valeur manuellement en éditant le fichier contenant la configuration atomique. Si c'est votre choix, assurez-vous de le faire dans un format qui contient les positions atomiques en coordonnées cartésiennes, comme le format XSF de XCrySDen, ou le format de données d'entrée de LAMMPS (et pas en coordonnées réduites, comme un fichier CFG par exemple). Par exemple, dans un fichier XSF, modifiez la troisième valeur dans le premier vecteur de boîte (affichée ici en gras) :
# Fcc Al oriented X=[1-12] Y=[-111] Z=[110].
CRYSTAL
PRIMVEC
198.21270999 0.00000000 1.43047700
0.00000000 140.15755135 0.00000000
0.00000000 0.00000000 2.86095404
CONVVEC
198.21270999 0.00000000 1.43047700
0.00000000 140.15755135 0.00000000
0.00000000 0.00000000 2.86095404
PRIMCOORD
(...)
ⓘ Selon le format de fichier, la valeur peut se situer à différents endroits. Veuillez vous référer à la documentation du code de simulation ou de visualisation correspondant au format de fichier utilisé.
Après avoir introduit cette inclinaison, la périodicité est restaurée suivant X. Toutefois, cette solution ne restaure pas la périodicité suivant Y. Les atomes proches des bords de boîte suivant Y devraient donc toujours être figés pour éviter des effets indésirables.
ⓘ La boîte inclinée telle que nous l'avons construite peut ne pas être acceptée par tous les codes de simulation. Par exemple, LAMMPS requiert que le premier vecteur soit aligné avec l'axe X, il n'autorise pas que ce vecteur soit incliné. Pour satisfaire cette exigence, vous pouvez utiliser l'option "-alignx" de Atomsk, ou introduire la dislocation suivant un axe différent, qui peut être incliné dans LAMMPS. Vous pouvez lire ce tutoriel, et vous référer à la documentation de LAMMPS pour plus d'information.
Parfois, il est préférable de construire un dipôle, ou un quadripôle de dislocations. Cela peut être réalisé avec Atomsk, en appelant l'option "-dislocation" plusieurs fois. Chaque fois que cette option est appelée, elle introduit une nouvelle dislocation dans le système.
Construisons un quadripôle de dislocations vis dans un système d'aluminium :
atomsk --create fcc 4.046 Al orient [1-12] [-111] [110] \
-duplicate 40 30 1 \
-dislocation 0.251*box 0.251*box screw Z Y 2.860954 \
-dislocation 0.751*box 0.251*box screw Z Y -2.860954 \
-dislocation 0.251*box 0.751*box screw Z Y -2.860954 \
-dislocation 0.751*box 0.751*box screw Z Y 2.860954 \
Al_quadrupole.cfg
Comme précédemment, nous créons une maille élémentaire d'aluminium avec le mode "--create", puis la dupliquons pour former une super-cellule. Ensuite, les quatre dislocations sont introduite deux avec un vecteur de Burgers positif aux coordonnées réduites (0.25,0.25) et (0.75,0.75), et deux avec un vecteur de Burgers négatif en (0.75,0.25) et (0.25,0.75). Chaque coordonnée est décalée d'une petite quantité (0.001) pour éviter que les dislocations ne soient placées exactement sur la position d'un atome.
Lorsque plusieurs dislocations sont introduites, leurs contributions aux composantes de contraintes s'ajoutent. Comme précédemment, il est possible de les visualiser, par exemple la composante σxz ressemble à la figure suivante :
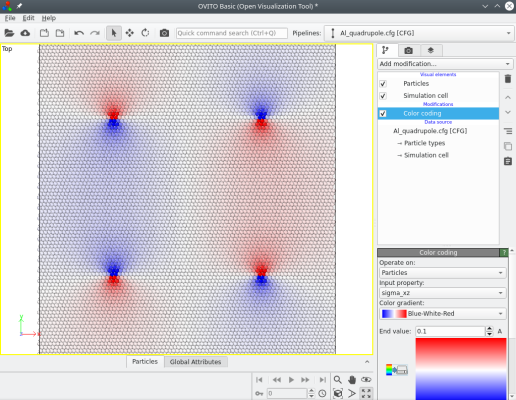
Cette fois, en bord de boîte, les déplacements dus aux quatre dislocations tendent à s'annuler (car la somme de leurs vecteurs de Burgers est un vecteur nul). Aucun bord n'a besoin d'être figé, il est possible d'appliquer des conditions aux limites périodiques dans les trois dimensions.
Au lieu d'introduire la dislocation suivant Z, il est possible de l'aligner avec la direction X ou Y. Pour ce faire, la maille élémentaire doit être orientée différemment dès le départ, et les directions données dans l'option "-dislocation" doivent aussi être modifiées. Comme exercice, vous pouvez essayer d'introduire la même dislocation vis que ci-dessus, mais dans la direction X ou Y.
Les dislocations construites avec les méthodes ci-dessus ne sont pas relaxées ni optimisées. Elles correspondent, au mieux, aux champs de déplacements prédits par la théorie élastique des dislocations. Toutefois, afin d'obtenir la véritable configuration atomique des dislocations, il sera nécessaire de réaliser une simulation avec une méthode ab initio ou un potentiel inter-atomique. Par exemple, dans le cas de l'aluminium, on s'attend à ce qu'une dislocation ½[110] se dissocie en deux dislocations partielles de Shockley. En fonction de la méthode choisie ci-dessus, la dislocation peut interagir avec des bords figés, l'empêchant de se dissocier pleinement et introduisant des effets indésirables. Vous aurez peut-être à tester différentes tailles de boîtes, et différentes méthodes, avant d'être sûr que vos résultats soient cohérents.
Ce tutoriel s'est focalisé sur les dislocations à caractère purement vis, il est aussi possible de construire des dislocations à caractère coin comme le montre le tutoriel précédent.
Par défaut, l'option "-dislocation" applique les champs de déplacement de l'élasticité isotrope. Cela se justifie dans le cas de l'aluminium, qui est un matériau relativement isotrope. Dans les matériaux où l'anisotropie est très prononcée, il peut être préférable d'utiliser les équations de l'élasticité anisotrope, comme expliqué dans ce tutoriel.