Atomsk
The Swiss-army knife of atomic simulations

The Swiss-army knife of atomic simulations




This tutorial explains how Atomsk deals with the box, the atom positions, and the boundary conditions.
For any atomic system, Atomsk works with at least two irreducible types of data:
Some systems may contain additional information (like the force or velocity of atoms), but an atomic system must always contain at least the three box vectors and the positions and atomic number of atoms. By default, Atomsk works with the Cartesian positions of atoms, which are absolute and independent of the box vectors.
Atomsk treats the box vectors as full vectors, i.e. each box vector can have any arbitrary component along the Cartesian X, Y and Z directions, and they can form any arbitrary angle (i.e. they are not required to be orthogonal). In other words, in the most general case the box vectors are H1=(x1 y1 z1), H2=(x2 y2 z2), H3=(x3 y3 z3).
This is worth mentioning, because it is not the case of all visualization or simulation softwares. For instance, LAMMPS requires that the first box vector has y1=z1=0, and the second box vector has z2=0. This means that it is impossible to write some vector components into a LAMMPS data file, and that it may be necessary to rotate the system before writing such a file. To know more you may read this tutorial.
Another example is the native format of VESTA, where the box vectors are saved using the "conventional cell" notation: only the lengths of the box vectors a, b, c, and the angles between them α, β, γ, can be written in a *.vesta file. So, when displaying the system, VESTA always assumes that the first box vector is aligned with the Cartesian X direction, and that the second one is contained within the XY plane. Thus, when writing files in this format, any information about a rotation of the box is lost.
Within Atomsk, the origin of the box is always the origin of Cartesian coordinates (0 0 0). Again, this is not the case for all simulation or visualization softwares. Some softwares (like LAMMPS) may shift the origin of the box (e.g. in LAMMPS, it takes the values xmin, ymin, zmin). When Atomsk reads such a file, the whole system (i.e. box and atom positions) are shifted, so that the origin of the box is placed at (0 0 0).
Atomsk works with the Cartesian coordinates of atoms (usually in Å), and treats atoms separately from the box. Atomsk does not automatically apply periodic boundary conditions: the atoms can have any (x y z) coordinates, even if that means that they are out of the box.
For the sake of example, let us create a unit cell of aluminium:
atomsk --create fcc 4.046 Al aluminium.xsf
If you open the file "aluminium.xsf" with a text editor, you will see that it contains the three box vectors, as well as the Cartesian positions of atoms (in Å):
# Fcc Al oriented X=[100], Y=[010], Z=[001].
CRYSTAL
PRIMVEC
4.04600000 0.00000000 0.00000000
0.00000000 4.04600000 0.00000000
0.00000000 0.00000000 4.04600000
CONVVEC
4.04600000 0.00000000 0.00000000
0.00000000 4.04600000 0.00000000
0.00000000 0.00000000 4.04600000
PRIMCOORD
4 1
13 0.00000000 0.00000000 0.00000000
13 2.02300000 2.02300000 0.00000000
13 0.00000000 2.02300000 2.02300000
13 2.02300000 0.00000000 2.02300000
In this file, all atoms are inside the box. Now, what happens if we apply a large displacement to all atoms, so that they end up out of the box? Let us try it with the option "-shift":
atomsk aluminium.xsf -shift 10 10 10 Al_shift.xsf
Now, the file looks like this:
# Fcc Al oriented X=[100], Y=[010], Z=[001].
CRYSTAL
PRIMVEC
4.04600000 0.00000000 0.00000000
0.00000000 4.04600000 0.00000000
0.00000000 0.00000000 4.04600000
CONVVEC
4.04600000 0.00000000 0.00000000
0.00000000 4.04600000 0.00000000
0.00000000 0.00000000 4.04600000
PRIMCOORD
4 1
13 10.00000000 10.00000000 10.00000000
13 12.02300000 12.02300000 10.00000000
13 10.00000000 12.02300000 12.02300000
13 12.02300000 10.00000000 12.02300000
Indeed, all coordinates of atoms were shifted by 10 Å.
This behavior is important: Atomsk keeps the Cartesian coordinates of the atoms. It does not automatically apply periodic boundary conditions (PBC), nor bring the atom back into the box. Keep that in mind when you construct your system: when atoms move around and exit the box, do not expect Atomsk to apply PBC automatically!
If you wish to apply PBC, you have to explicitely ask for it, with the option "-wrap" as explained in section 4 below.
The visualization of systems containing atoms that are out of the box can be tricky. Let us convert the atomic system (the shifted one) into CFG format for visualization:
atomsk Al_shift.xsf cfg
This will produce a file named "Al_shift.cfg". Let us visualize it with OVITO, as exampled below:
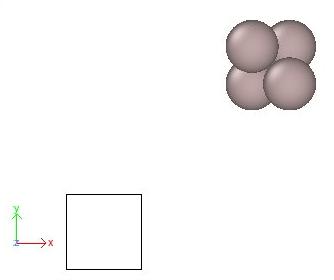
OVITO displays the Cartesian base (coloured arrows labeled x, y, z), the simulation box (black lines), and the atoms as spheres. Indeed, the atoms are outside of the box, as expected from the commands that we ran above.
Now, let us visualize the exact same file ("Al_shift.cfg") with Atomeye:
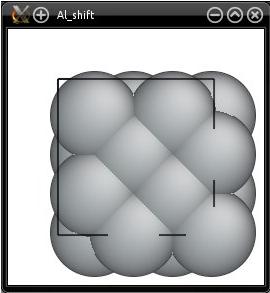
Two things seem to be wrong here. First, Atomeye displays atoms as if they were inside of the box. Why is that? This is because Atomeye automatically applies periodic boundary conditions, and places all atoms inside the box. The second thing is that there are only four atoms in our system, but Atomeye displays more atoms than that. This is because Atomeye considers that our system is "too small", so it duplicates it twice in each direction, as explained in the previous tutorial.
In summary, it is important that you know the limitations and caveats of the visualization softwares that you use. Some visualization softwares do not show the actual coordinates of atoms as they are written in the file. Instead, they automatically (and silently) apply periodic boundary conditions. Sometimes it may be desirable to ease the visualization, and sometimes it can be misleading in interpreting your data. So, be careful when visualizing your atomic systems. Read the documentation of the software to know what boundary conditions they apply, and if they perform other operations silently.
As stated earlier, Atomsk does not automatically apply periodic boundary conditions, nor put atoms back into the box. If you want all atoms to be inside the box, you have to ask for it explicitely with the option "-wrap". For instance, let us read the file "Al_shifted.cfg" constructed earlier, where all atoms are out of the box, and let us wrap them:
atomsk Al_shift.cfg -wrap Al_wrap.cfg
The option "-wrap" will shift all atoms using the box vectors, until their coordinates fall into the box. Visualization with OVITO confirms that atoms are now indeed inside the box:
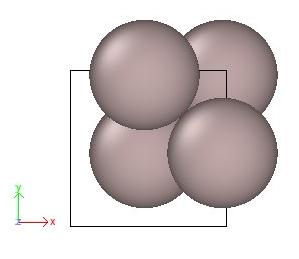
It is possible to tell Atomsk to convert all atom coordinates from Cartesian (the default) into reduced (or fractional) coordinates, thanks to the option "-fractional":
atomsk Al_shift.xsf -fractional Al_frac.xyz
Reduced, or fractional, coordinates are expressed as a fraction of the box vectors. If atoms are inside the box, all of their coordinates have values between 0 and 1. However, if an atom is out of the box, its reduced coordinates can be smaller than 0 or greater than 1.
Remember that the option "-fractional" does not wrap atoms back into the box, it just divides their coordinates by the box vectors.
As stated earlier, Atomsk always required that the box vectors, as well as the positions and atomic numbers of atoms, are defined. This is the absolute minimum. Then, other information may exist.
Sometimes, the input file contains additional information about atoms. For instance, a LAMMPS dump file may contain the atom "type", or the force it is submitted to, or its total energy, and so on. In such a case, Atomsk also stores all these additional information, so that it can be transferred to the output file, if relevant. However, note that some file formats do not support such additional properties, in which case Atomsk will not write them, and some properties will be lost.
In some simulations, an atomic system may also contain ionic shells. Atomsk also stores the positions of shells, and most options (like "-duplicate", "-shift", and so on) are compatible with shells. You may read this tutorial to know more about how Atomsk treats ionic cores and shells.
When you construct a system with Atomsk, you may shift atoms, insert a dislocation, or apply other transformations that may displace some atoms out of the box. You should keep in mind that Atomsk does not automatically apply periodic boundary conditions, so depending on the transformations (options) that you apply, some atoms may exit the box.
If you want to make sure that all atoms are inside the box, simply use the option "-wrap".