Atomsk
The Swiss-army knife of atomic simulations

The Swiss-army knife of atomic simulations




This tutorial explains how to use Atomsk to orient unit cells, and duplicate them to create supercells, using silicon as an example.
Let us start with the most basic configuration: create a unit cell of silicon. This can be achieved very easily by executing Atomsk with the mode --create:
atomsk --create diamond 5.431 Si xsf
In this case the output file will be "Si.xsf". It can be visualized with XCrySDen or with VESTA:
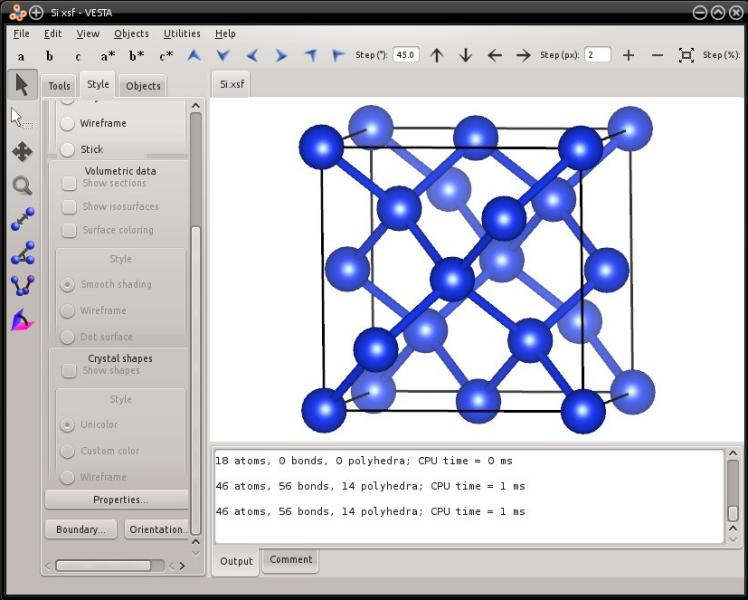
Now, Atomsk offers the possibility to generate a unit cell with a different orientation. For instance, to generate the unit cell corresponding to the lattice orientation X=[-110], Y=[001], Z=[110], the syntax is the following:
atomsk --create diamond 5.431 Si orient [-110] [001] [110] xsf
Note that the instruction "orient" must be specified immediately after the atom species "Si". The unit cell then looks like the following:
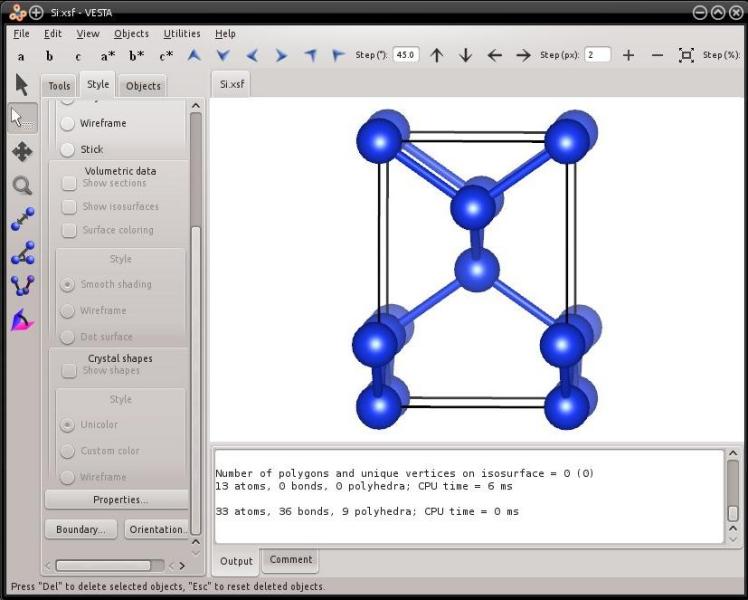
You may try to generate different crystal orientations. Note that the three directions defined with Miller indices have to be normal to each other, otherwise Atomsk will produce an error.
Now that we know how to generate a unit cell with the desired crystal orientation, let us duplicate it and generate what is called a supercell. This can be achieved with the option -duplicate, followed by the number of repetitions along each crystal direction. The number of repetitions must be an integer. If you use "-duplicate 1 1 1", then the system remains identical (because it is multiplied by 1 along each direction).
Let us use the unit cell "Si.xsf" that was generated before. In order to duplicate it three times along the X direction, use:
atomsk Si.xsf -duplicate 3 1 1 Si_supercell.xsf
This will produce the file "Si_supercell.xsf" containing the supercell. The system looks like:
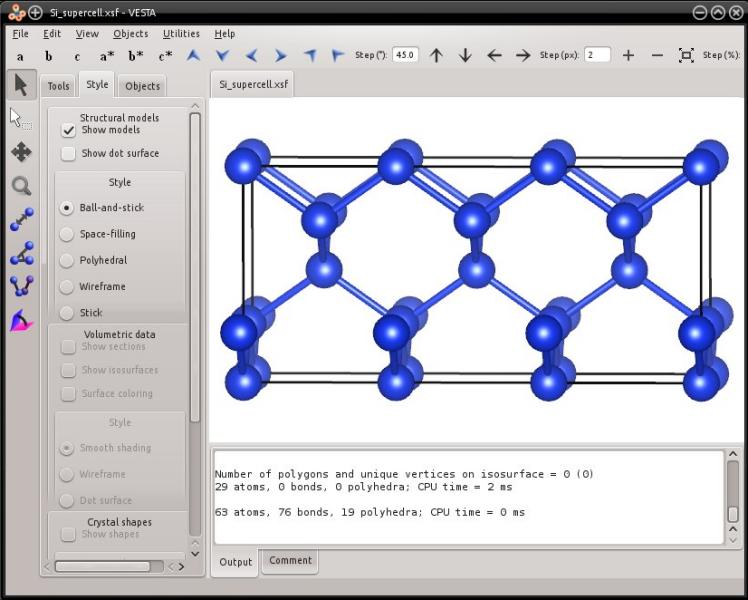
Now you may also duplicate it several times along each direction of space to generate even larger systems, for instance "-duplicate 10 6 6":
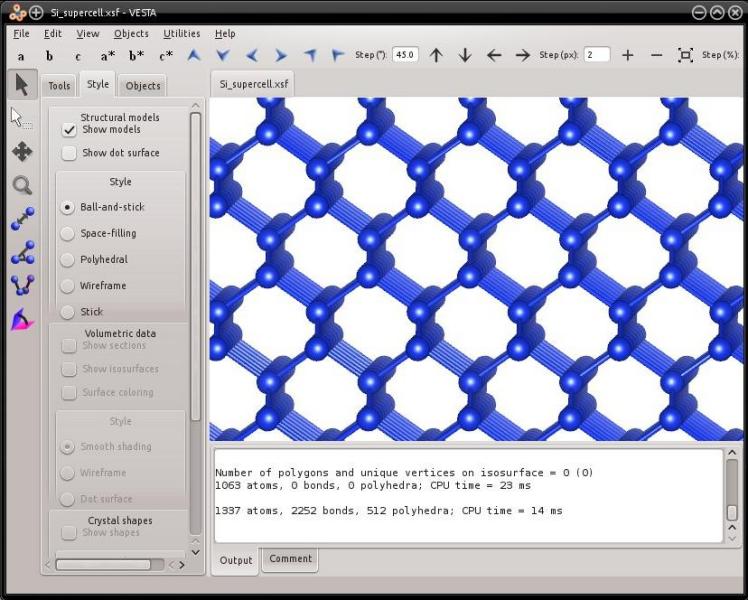
In the example above, we first created a file named "Si.xsf" containing the unit cell, and then duplicated it and wrote the supercell into another file. There is a faster way to do this, by combining the mode "--create" with the option "-duplicate":
atomsk --create diamond 5.431 Si orient [-110] [001] [110] -duplicate 3 1 1 Si_supercell.xsf
With this single command, Atomsk will create the oriented unit cell, then duplicate it, and write only one final file containing the supercell.
Of course, one or several options can be used. You may duplicate, cut, rotate the system, insert a dislocation, etc., all inside a single command.
Orienting and duplicating unit cells is pretty easy with Atomsk. As an exercise, you can use the unit cells generated in the previous tutorial, for instance aluminium or iron, and orient and duplicate them.