Atomsk
Le couteau suisse pour les simulations atomiques

Le couteau suisse pour les simulations atomiques




Dans ce tutoriel, vous allez apprendre à générer un joint de grains entre deux cristaux, souvent appelé bicristal. D'abord nous couvrirons les joints de grains de faible indice, puis les joints symmétriques avec un angle de désorientation arbitraire. Enfin nous discuterons le cas des joints de grains généraux.
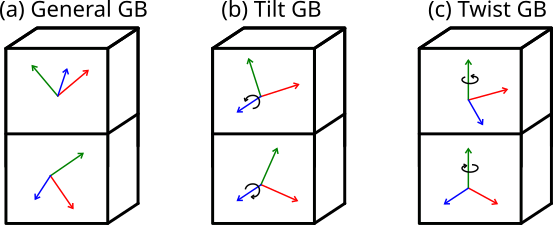
Un joint de grains est l'interface entre deux cristaux, ou cristallite, dont les orientations cristallines diffèrent. Dans le cas le plus général, la désorientation est arbitraire, comme illustré sur la Fig. (a) ci-dessous. Quand les deux cristaux partagent un même axe de rotation parallèle au plan du joint, comme sur la Fig. (b), on parle de joint de flexion (tilt). Quand ils partagent un axe de rotation normal au joint comme dans la Fig. (c), on parle de joint de torsion (twist). Quand les angles de rotation sont exactement opposés, de sorte que les deux grains ont la même périodicité dans le plan du joint, alors le joint de grains est dit symétrique.
ⓘ Avant d'utiliser Atomsk, il est important de concevoir votre système « sur papier ». Déterminez les directions cristallines et/ou les rotations nécessaires pour atteindre votre but -sans quoi Atomsk ne vous sera d'aucune aide.
Pour commencer, considérons deux cristaux d'aluminium cfc tournés autour de leur axe [001], puis collés suivants leurs plans {210}. Remarquez que d'un côté de cette interface il y aura le plan (210) d'un cristal, et de l'autre côté le plan (210) de l'autre crystal. Un tel joint de grains est un joint de flexion symétrique {210}[001].
Puisque les deux directions cristallines sont connues, le plus simple est d'utiliser le mode "--create" pour générer les deux cristaux avec l'orientation voulue. Ici nous utilisons Z=[001] comme axe de rotation, la normale au plan du joint est Y=[210], et par déduction la troisième direction doit être X=[120] :
atomsk --create fcc 4.046 Al orient [-120] [210] [001] crystal1.cfg
Maintenant nous faisons la même chose pour le second cristal, en inversant la direction cristalline suivant Y pour qu'elle devienne Y=[210] :
atomsk --create fcc 4.046 Al orient [-120] [-2-10] [001] crystal2.cfg
Il ne reste plus qu'à empiler les deux cristaux suivant la direction Y pour former le bicristal :
atomsk --merge Y 2 crystal1.cfg crystal2.cfg Al_210_001.cfg
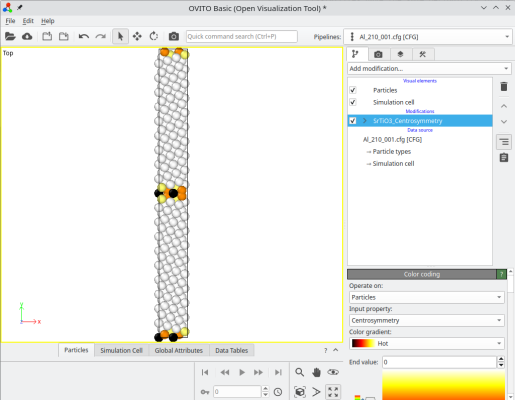
Le système final peut être visualisé, par exemple avec OVITO. En accolant les deux systèmes, Atomsk crée une nouvelle propriété auxiliaire appelée "sysID", indiquant pour chaque atome à quel système il appartenait, dans ce cas sysID=1 pour le premier cristal, et sysID=2 pour le second. Cete propriété "sysID" peut être visualisée en utilisant un code couleur (deuxième image ci-dessous). Enfin, vous pouvez aussi visualiser les défauts en activant une analyse, par exemple le critère de centro-symétrie, pour mettre en évidence le joint de grains.
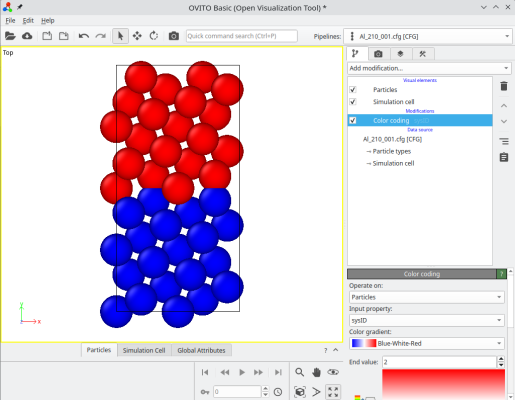
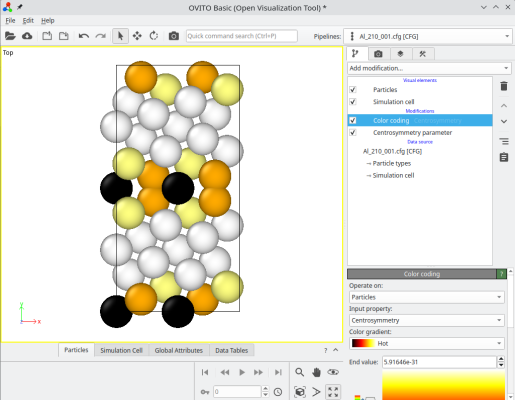
ⓘ En empilant les deux cristaux, il existe un joint de grains au milieu de la boîte finale. Si vous utilisez des conditions aux limites périodiques (comme montré ici dans OVITO), alors il existe aussi un second joint en bord de boîte suivant Y (voir les atomes colorés en haut et en bas de la boîte). Pour un joint de grains symétrique avec un réseau cubique, ces deux joints sont équivalents. Cependant, si le joint n'est pas symétrique, ou si le réseau n'est pas cubique, alors les deux joints peuvent avoir des structures différentes, et donc des propriétés différentes.
Bien sûr, un tel système est probablement trop petit pour réaliser des calculs, les deux joints de grains sont trop proches l'un de l'autre. Pour palier cela, vous pouvez simplement utiliser l'option "-duplicate" pour créer les deux cristaux initiaux, par exemple pour dupliquer cinq fois suivant Y :
atomsk --create fcc 4.046 Al orient [-120] [210] [001] -duplicate 1 5 1 crystal1.cfg
atomsk --create fcc 4.046 Al orient [-120] [-2-10] [001] -duplicate 1 5 1 crystal2.cfg
atomsk --merge Y 2 crystal1.cfg crystal2.cfg Al_210_001.cfg
Maintenant chaque grain est plus épais suivant Y, de sorte que la distance entre les joints de grains est plus grande :
Selon ce que vous voulez faire, vous pouvez aussi dupliquer le système suivant les directions X et Y du joint. Si vous avez besoin de déplacer un grain par rapport à l'autre, vous pouvez sélectionner les atomes appartenant à ce grain (sysID=2), et leur appliquer l'option "-shift" :
atomsk --merge Y 2 crystal1.cfg crystal2.cfg Al_210_001.cfg -select prop sysID 2 -shift 4 0 0
Ainsi les atomes du second grain sont bien déplacés :
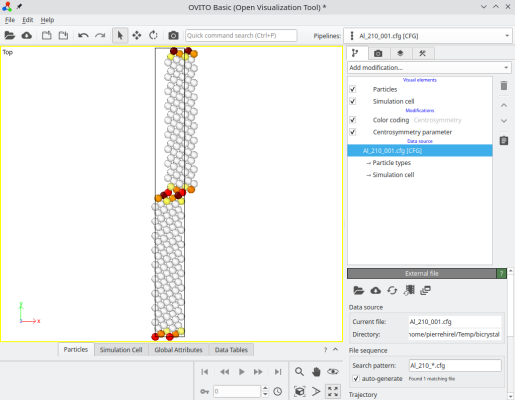
ⓘ Vous pouvez ajouter l'option "-wrap" si vous voulez que tous les atomes soient dans la boîte.
Pour vous exercer avec les commandes ci-dessus, vous pouvez essayer de construire différents joints de grains : {310}[001], {410}[001]. Vous pouvez aussi essayer avec des matériaux différents, par exemple du fer (cubique centré), ou un oxyde comme MgO (structure NaCl ou rock-salt).
Tournons-nous vers un autre type particulier de joints de grains, un joint de torsion. Cette fois, nous plaçons l'axe de rotation [001] suivant la direction Cartésienne Y, et tournons les deux cristaux autour de cet axe avec des angles opposés, de sorte que l'axe [210] d'un grain soit aligné avec l'axe [210] de l'autre. Nous dupliquons aussi suivant Y pour éviter que le système ne soit trop petit :
atomsk --create fcc 4.046 Al orient [-120] [001] [210] -duplicate 1 5 1 crystal1.cfg
atomsk --create fcc 4.046 Al orient [-120] [001] [-2-10] -duplicate 1 5 1 crystal2.cfg
atomsk --merge Y 2 crystal1.cfg crystal2.cfg Al_twist_001.cfg
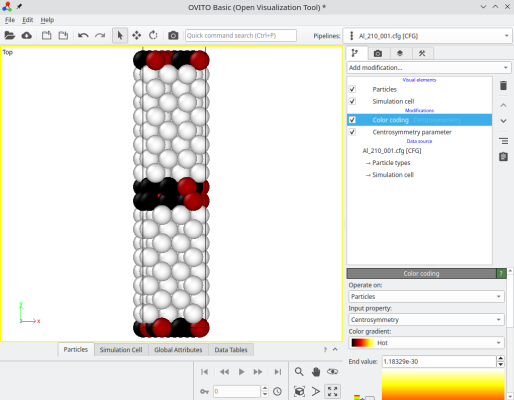
Là encore, vous pouvez essayer avec des orientations cristallines différentes, et/ou avec différents matériaux.
Les sections précédentes ont décrit des joints de grains particuliers, correspondant à des directions de faibles indices de Miller, comme {210}, {310}, etc. Et si à la place, nous voulons créer un joint de désorientation α arbitraire ?
Pour construire un joint de grains avec une désorientation arbitraire, vous pouvez utiliser l'option "-rotate", suivie de l'option "-orthogonal-cell". Par exemple, pour tourner les cristaux de ±5.1°:
atomsk --create fcc 4.046 Al -rotate Z 5.1 -orthogonal-cell crystal1.cfg
atomsk --create fcc 4.046 Al -rotate Z -5.1 -orthogonal-cell crystal2.cfg
atomsk --merge Y 2 crystal1.cfg crystal2.cfg Al_51.cfg
Après avoir tourné le cristal, l'option "-orthogonal-cell" recherche des combinaisons linéaires des vecteurs du réseau cristallin, qui produisent de nouveaux vecteurs alignés avec les axes cartésiens X, Y, et Z. Cela permet de s'assurer que les nouveaux vecteurs de boîte respectent la périodicité du réseau cristallin, de sorte que le système reste périodique suivant X, Y, et Z. Parfois, cette nouvelle boîte orthogonale est beaucoup plus grande que la boîte initiale, dans cet exemple elle compte environ 12 600 atomes. Ici nous avons construit un joint de flexion symétrique, où chaque cristal est tourné de ±5.1°, produisant une désorientation de 10.2° entre les deux grains. Le système final peut être visualisé avec OVITO :
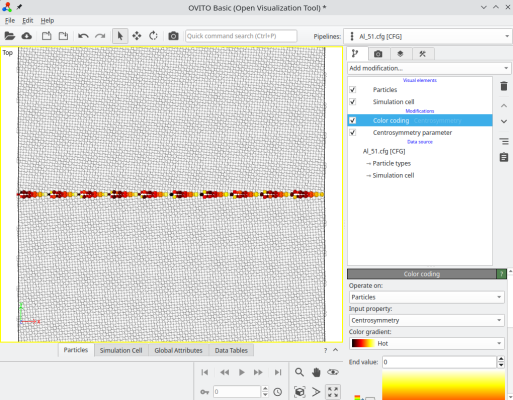
ⓘ Pour certains angles, ou si le réseau n'est pas cubique, il est possible que l'option "-orthogonal-cell" échoue à trouver de nouveaux vecteurs de boîte.
Pour produire un joint de torsion, le principe est le même, il suffit de tourner le système autour de Y. Seulement cette fois, nous devons aussi dupliquer le système suivant Y pour éviter qu'il ne soit trop petit :
atomsk --create fcc 4.046 Al -rotate Y 5.1 -orthogonal-cell -duplicate 1 10 1 crystal1.cfg
atomsk --create fcc 4.046 Al -rotate Y -5.1 -orthogonal-cell -duplicate 1 10 1 crystal2.cfg
atomsk --merge Y 2 crystal1.cfg crystal2.cfg Al_51.cfg
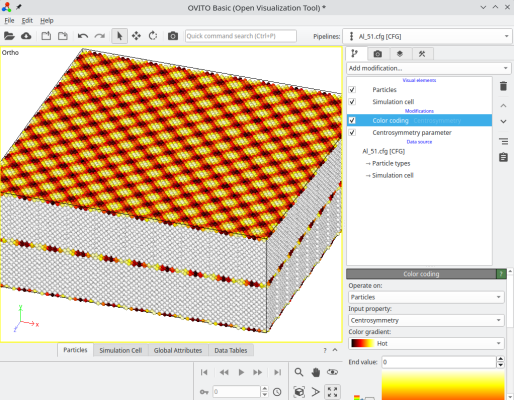
Suivant l'angle de rotation choisi, les systèmes construits de cette façon peuvent être assez grands.
Avec les méthodes présentées ci-dessus, il est assez simple de construire des joints de grains de flexion ou de torsion avec n'importe quelle désorientation donnée. Cependant cette construction n'est souvent qu'une première étape, gardez en tête que ces joints ne sont pas relaxés. Il est ensuite nécessaire de recourir à une simulation pour minimiser l'énergie. Souvent, il est nécessaire de calculer l'énergie en fonction du déplacement d'un grain (ce qu'on appelle une γ-surface) pour trouver le minimum (ou les minima) d'énergie.
Pour générer un polycristal, c'est-à-dire un système contenant plusieurs cristallites d'orientations différentes, consultez le tutoriel suivant.