Atomsk
The Swiss-army knife of atomic simulations

The Swiss-army knife of atomic simulations




After almost 3 years of development, the version beta 0.11 of Atomsk was released in early 2021. This version shows improved performance thanks to a re-writing of some routines, or thanks to parallelization with OpenMP. These improvements are presented below.
Major performance improvements were possible thanks to parallelization of the code with OpenMP. Now, Atomsk can create several threads that will use all the CPU cores in your machine.
What does it mean? Before (version beta-0.10.6 and earlier), Atomsk could run only one thread, meaning it could use only one CPU core on your machine. Most modern computers have a CPU with several cores. As an example, if your CPU has 4 cores, it means that Atomsk was using only 25% of your computer power. Now with the version beta-0.11, Atomsk can use 100% of your computer power, meaning that it will run faster.
Note that only some specific parts of the code are actually parallelized. The vast majority of the code is not, so don't expect your CPU to be used 100% every time you use Atomsk, but only when you perform some specific operations.
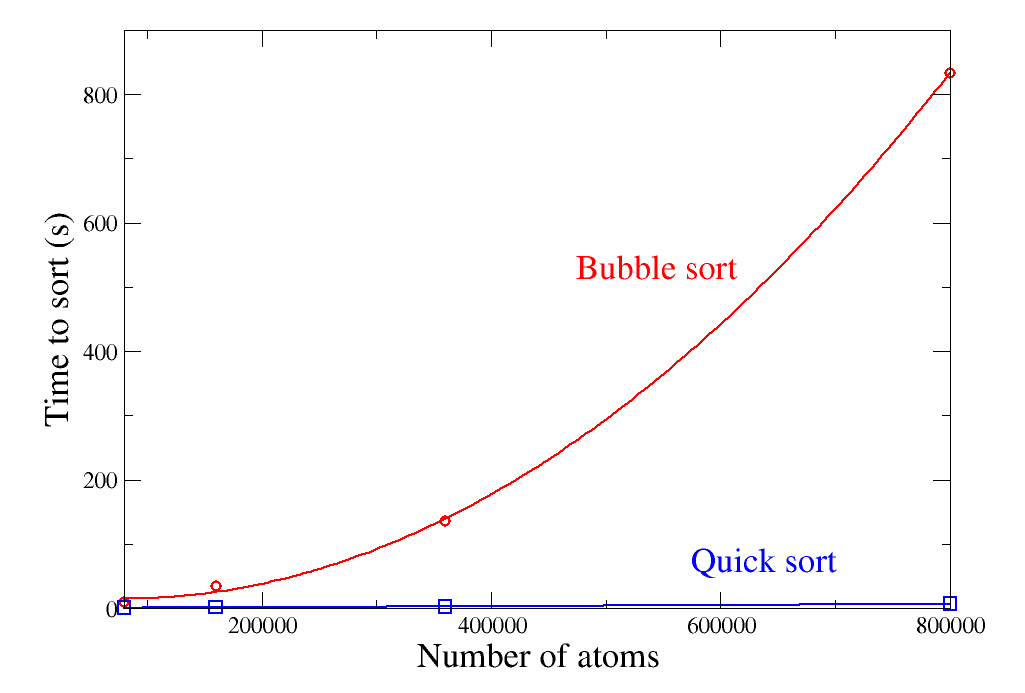
For a long time, Atomsk relied on the "bubble sort" algorithm to sort arrays like a list of atoms. This algorithm is one of the most simple to implement, but also very inefficient, scaling as N2 where N is the number of atoms. Since the version beta-0.9 Atomsk uses the Quicksort algorithm, which is mush faster and scales as N. Although not a new addition in the version beta-0.11, this performance gain is noteworthy. The sorting algorithm is used in options "-sort" and "-select".
The option "-orthogonal-cell" was introduced in version beta-0.10. It allows converting a cell of any arbitrary geometry into an orthogonal cell, while preserving the crystal symmetry. To achieve that, it considers linear combinations of the initial cell vectors, and tries to find combinations that are aligned with the Cartesian X, Y and Z axes.
As implemented initially, this option was not very effective. Often it could not find a suitable orthogonal cell, or was taking forever to fill the new cell with atoms. This is now improved in version beta 0.11: not only the algorithm is more robust and should find an orthogonal cell more easily, but it is also parallelized with OpenMP for faster results.
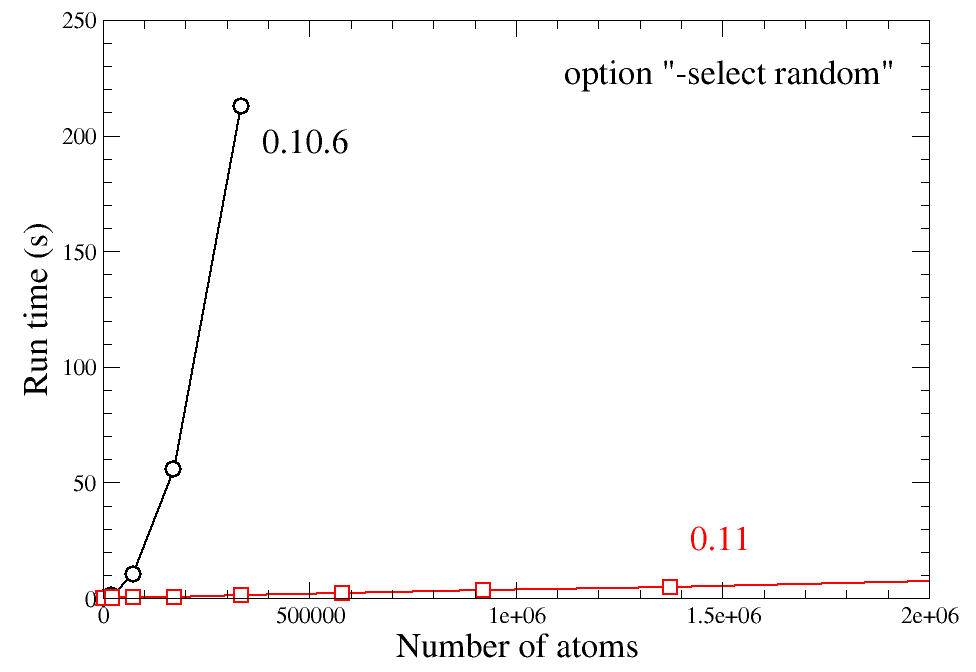
The option "-select" is one of the most versatile in Atomsk. It allows selecting atoms depending on various criteria, and to combine several selections (see this tutorial).
One of the possibilities is asking Atomsk to select a given number of atoms at random. In previous versions (beta-0.10.6 and earlier), this option performed very poorly, with an execution time scaling exponentially with the number of atoms. This made this option almost impossible to use in systems containing more than a few 100,000 of atoms.
The algorithm for selecting random atoms was completely changed, which considerably improves its performance. The new algorithm scales linearly as function of the number of atoms N, and is incomparably more efficient in systems with millions of atoms.
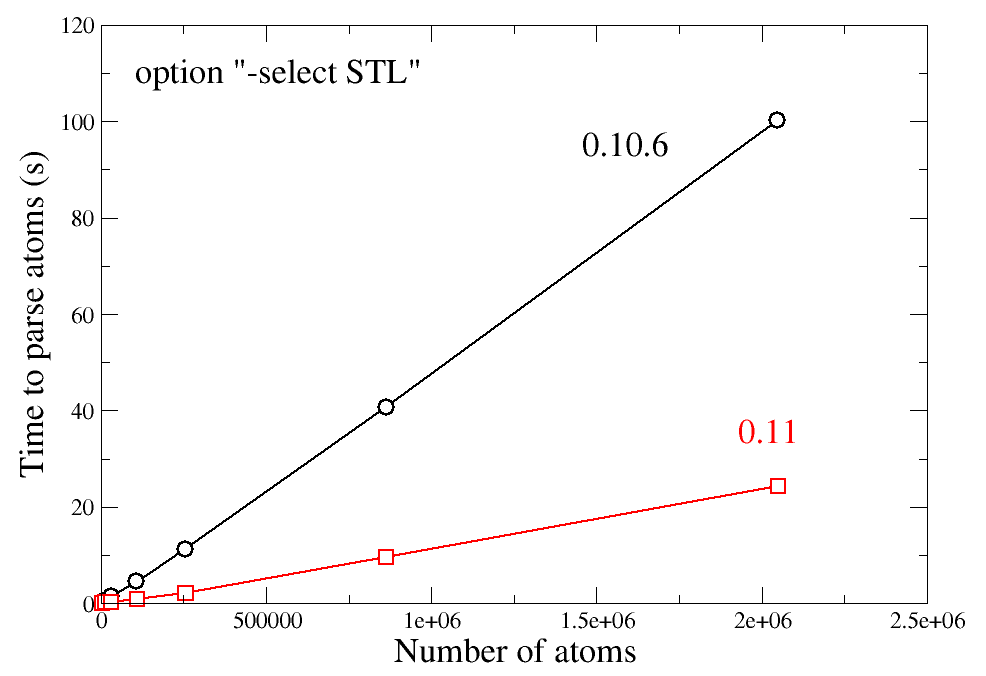
Another possibility of the option "-select" is to select atoms according to a 3-D shape saved in a file with the STL format (see this tutorial). In version beta-0.11, this routine was parallelized using OpenMP: it can now take advantage of all the cores of your CPU. The graph below compares the ancient version beta-0.10.6 which was not parallelized (1 thread), with the new version beta-0.11 running on 8 cores.
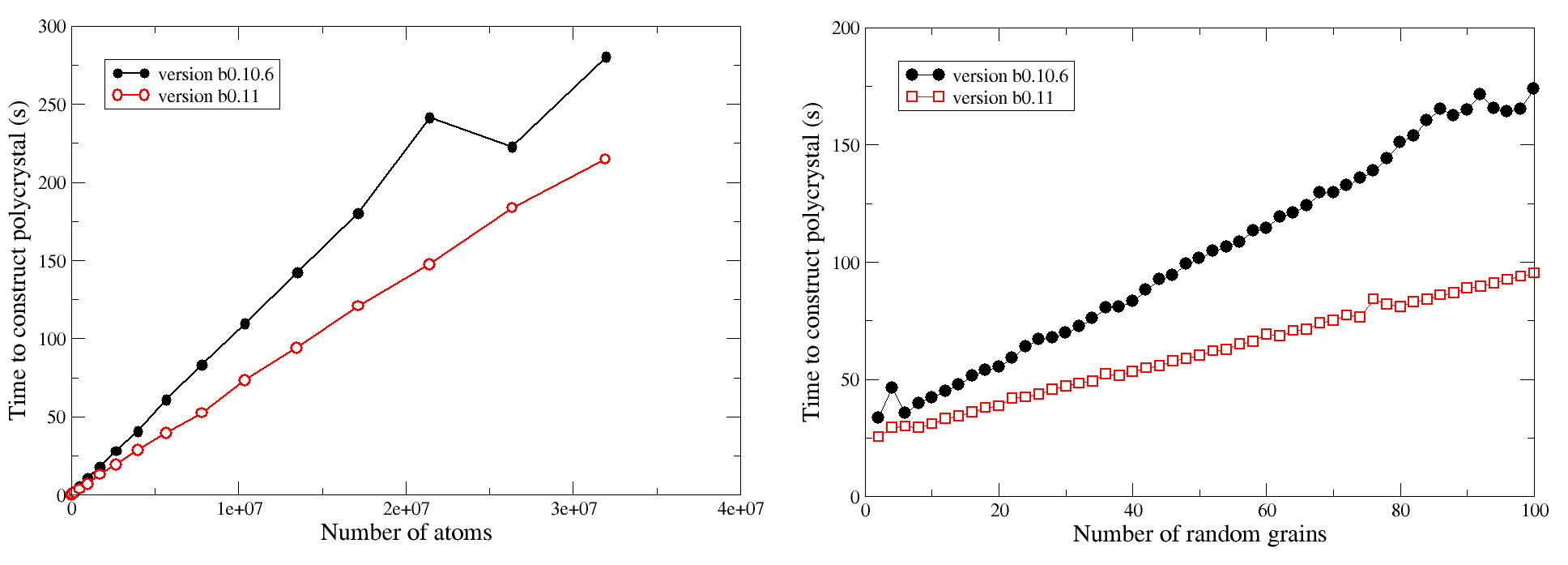
Based on the articles citing Atomsk, the mode "--polycrystal" appears to be one of the most popular. It allows generating polycrystals from any given initial structure, whether it is a crystal unit cell, or a cell containing twins, dislocations or other defects.
In the version beta-0.11 the mode "--polycrystal" was modified to be more robust. It was also parallelized with OpenMP, leading to better performances. Although the gain is not as impressive as with the previous options, users will certainly appreciate it. The first graph shows the time needed for Atomsk to construct a polycrystal with 12 grains, as function of the number of atoms (final cell size). The second graph shows the time as function of the number of grains, for a polycrystal containing 4 millions of atoms.