Atomsk
The Swiss-army knife of atomic simulations

The Swiss-army knife of atomic simulations




This tutorial gives examples to apply displacements or displacement functions to atoms.
▶ For more information, read the documentation of the options "-shift", "-rotate", "-select", and "-properties".
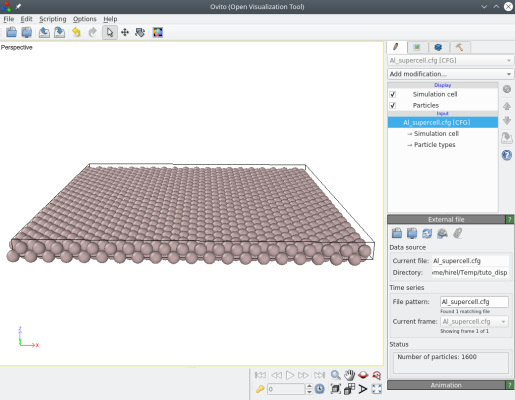
For this tutorial, we will use a supercell of aluminium:
atomsk --create fcc 4.046 Al -duplicate 20 20 1 Al_supercell.cfg
The option "-shift" is meant to easily translate atoms. It only requires the three Cartesian components of the translation vector, in angströms:
atomsk Al_supercell.cfg -shift 10.5 20.6 0.0 shift.cfg
With this command, all atoms are translated as can be visualized with OVITO:
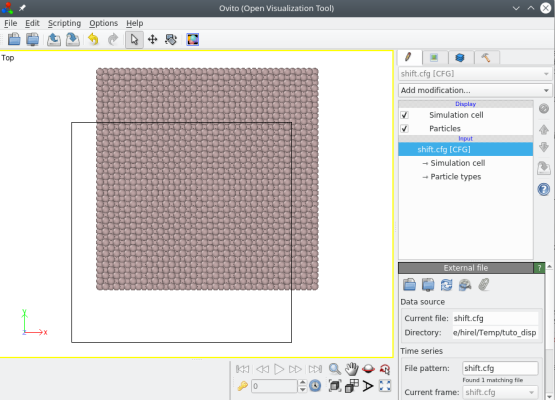
ⓘ Note that the box is not translated, because within Atomsk the origin of the box is always the Cartesian origin (0,0,0).
This option can also be used to translate only atoms that are above or below a given plane. For that, add the keyword "above" (or "below"), the distance and the normal to the plane, followed by the shift vector, for instance:
atomsk Al_supercell.cfg -shift above 40 Y 10.5 0 0 shift.cfg
Only atoms with a Y coordinate greater than 40 Å are displaced:
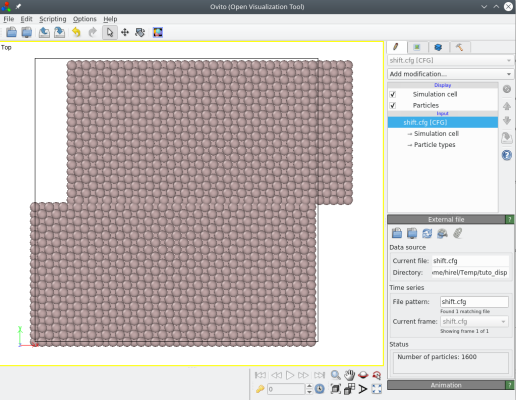
It is also possible to give the distance to the cut plane as an operation (for example "0.5*box"), or to give the normal to the plane as a crystal direction, like [110] or [-111]. Please refer to the documentation page of the option "-shift" for more information.
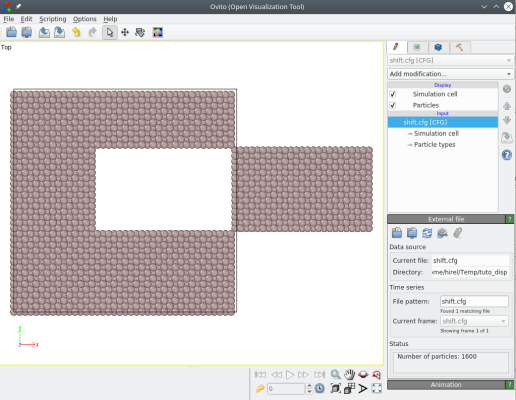
Finally, if the option "-select" is used to select part of the system, then only the selected atoms are translated. For example, let us select atoms in a box and translate them:
atomsk Al_supercell.cfg -select in box 30 30 -10 100 60 10 -shift 50 0 0 shift.cfg
You may also use the option "-select" to select one or several atoms according to their index. For example, if you wish to translate atoms number 45 and 86, you may use:
atomsk Al_supercell.cfg -select 45,86 -shift 50 0 0 shift.cfg
Please read the documentation of the option "-select" to discover the various possibilities.
The option "-rotate" allows to rotate the system around a given axis:
atomsk Al_supercell.cfg -rotate Z 15 rotated.cfg
The command above rotates the whole system around the Z axis by 15 degrees:
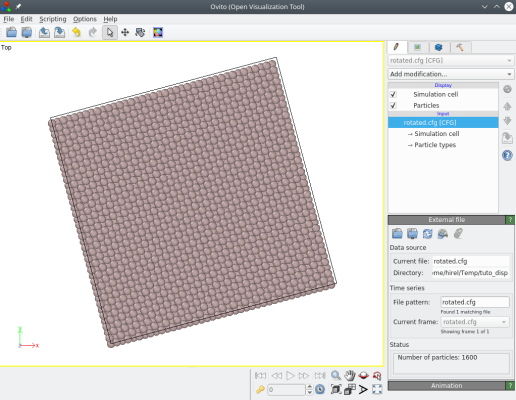
ⓘ Note that both the atoms and the box are rotated, so that the box vectors are not aligned with the Cartesian X and Y directions any more.
If you wish to rotate a group of atoms around its center of mass, then select them with the option "-select", and then use the option "-rotate" with the keyword "com". For example, let us rotate a ball of atoms at the center of the box:
atomsk Al_supercell.cfg -select in sphere 0.5*box 0.5*box 0.5*box 20 -rotate com Z 33 shift.cfg
Note that when a group of atoms is selected, then the box remains untouched (it is not rotated), only the selected atoms are rotated.
ⓘ It is possible to specify the position of the center of the sphere in angströms, or in reduced coordinates thanks to the keyword "box" as in the example above. However the radius of the sphere must be given as a number in angströms.
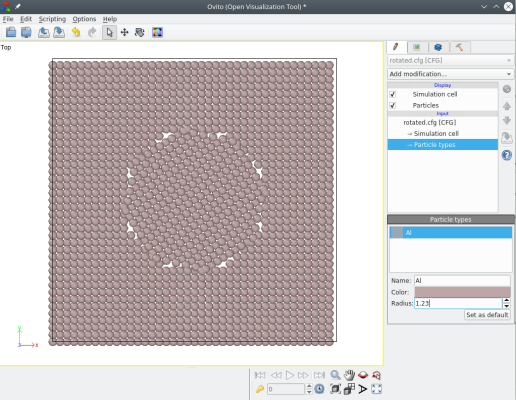
In addition to the three Cartesian axes (X, Y, or Z), the rotation axis can also be a Miller direction. Please refer to the documentation page of the option "-rotate".
The option "-disturb" applies random displacements to atoms. The displacements follow a normal distribution, and are adjusted so that there is no global translation of the system. The norms of the displacements are between zero (no displacement) and the maximum value that you provide.
For example, let us apply random displacements, with a maximum of 1.5 Å:
atomsk Al_supercell.cfg -disturb 1.5 random.cfg
Visualization with OVITO shows that Atomsk has applied random displacements (ux,uy,uz):
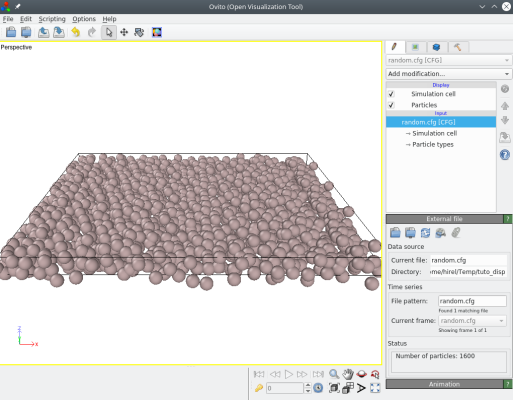
It is also possible to provide three values, corresponding to the maximum displacement along each Cartesian direction. For example, if we want to displace atoms only along Z we can run:
atomsk Al_supercell.cfg -disturb 0 0 1.0 random2.cfg
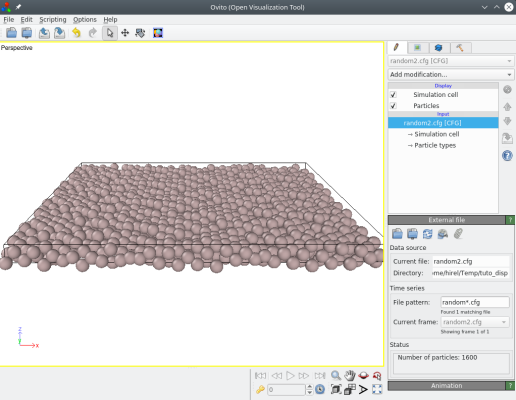
Again, you may try with various values and see the result.
If you wish to apply a specific displacement that is different for each atom, you may use the option "-properties". This option reads a file containing information about atoms. Here, we will use it to read displacements that we want to apply to atoms.
First, let us write the displacements in a text file. The file must begin with the keyword "displacement", followed by lines indicating the atom index, and the displacement dx dy dz to apply. Here is an example (lines starting with the # symbol are comments and are ignored by Atomsk):
# Atom displacements (in angströms)
displacement
1 0.2 0.2 2.5
2 -1.2 0.1 -2
3 -0.1 0.08 -1.3
30 -0.6 0.08 3.9
You do not have to write displacements for all atoms, nor to give them in any specific order. The file may contain displacements only for some atoms, and in any order -Atomsk will just apply each displacement to the corresponding atom with the given index. You can try adding more lines to the file above, to change the displacements or to displace other atoms.
Then, use the option "-properties" to read those displacements and apply them to the atomic system:
atomsk Al_supercell.cfg -properties disp.txt disp.cfg
The result can be visualized with OVITO, where we see that atoms were indeed displaced by Atomsk:
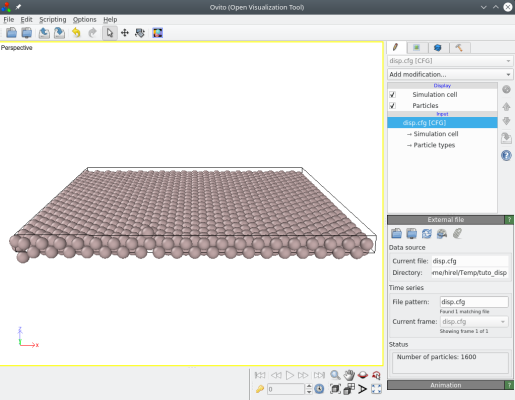
The option "-properties" allows displacing atoms according to a general formula. To achieve that, use the keyword "displacement function", and then give the displacement functions ux, uy and/or uz. Each function must start with "ux=" (or "uy=" or "uz=") followed by a function that may contain numbers, operators (+, -, *, /, ^), mathematical functions (cos, sin, tan, exp, sqrt...), or the constant "pi". Please refer to the documentation for a full description.
As an example, let us write a function for the displacements uz (i.e. along the Z direction):
# Atom displacements (in angströms)
displacement function
uz = 10*cos(4*pi*x/Hx)
The keyword "pi" will be automatically replaced by the value of π, Hx by the length of the first cell vector, and x by the coordinate of each atom. As before, use the option "-properties" to apply these displacements:
atomsk Al_supercell.cfg -properties disp.txt function.cfg
The resulting file "function.cfg" can be visualized with OVITO. Indeed, Atomsk has displaced atoms according to the given cosinus function:
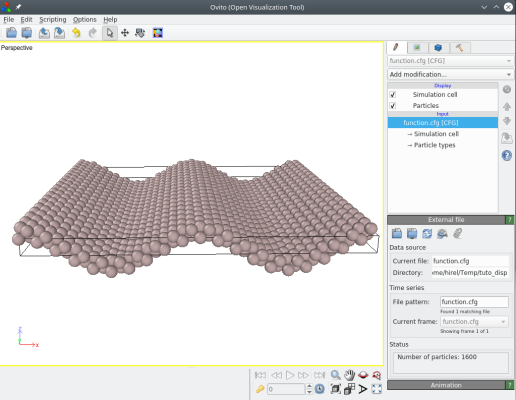
It is possible to modify the function, so that the displacement uz also depends on the y coordinate of atoms, for example:
# Atom displacements (in angströms)
displacement function
uz = 2*cos(4*pi*x/Hx) + 2*cos(4*pi*y/Hy)
Run the same command again:
atomsk Al_supercell.cfg -properties disp.txt function2.cfg
Then open the new file "function2.cfg" with OVITO (here atoms are coloured according to their Z coordinate to make displacements more visible):
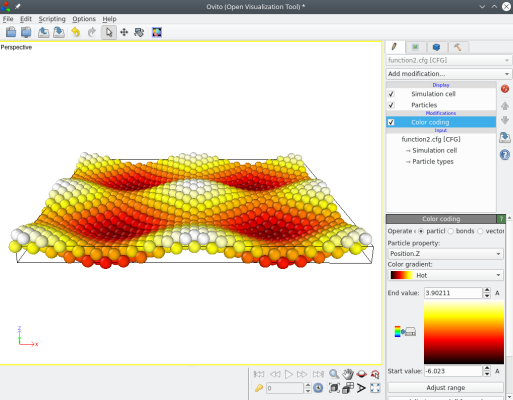
You may modify the function as you please, for example to change its periodicity: replace "4*pi" with "6*pi" or "2*pi", and visualize the result. You may also define displacement functions ux and uy. All you have to do is modify the text file, and then run the same command to apply it.
ⓘ For a list of supported mathematical functions, please read the documentation of the option "-properties".
A warning should be written here. This option may be slow, as Atomsk must evaluate the function for each atom. This option is parallelized, meaning it will use all the CPU cores on your machine for better efficiency. Still, if you write a very complicated function (with many operators, mathematical functions, parenthesis, etc.), then it may take a very long time for Atomsk to process it. In some cases it is possible that Atomsk can not interpret the function.
As it can be seen from the examples above, after applying a displacement some atoms may be out of the box. Note that Atomsk will not apply periodic boundary conditions automatically. If you want to do so, then you have to ask for it explicitely by adding the option "-wrap". However, beware that doing so may cause atoms to overlap.
In this tutorial we used a "flat" supercell of aluminium, because it makes it easier to visualize the displacements. Of course, the concepts described above can be used in different systems, for example a graphene sheet, or a larger 3-D system -the only limit is your imagination.