Atomsk
The Swiss-army knife of atomic simulations

The Swiss-army knife of atomic simulations




This tutorial explains how Atomsk reads CIF files.
The previous tutorials have illustrated how to create atomic systems with Atomsk, using the mode "--create". However, this mode only supports a very small set of lattice types, and is completely useless if you are interested in more complex structures.
The Crystallographic Information File (CIF for short) is a standard format, designed by the International Union of Crystallography. As such, it is a very powerful and broadly used format. CIF files for many compounds can be downloaded from the American Mineralogist Crystal Structure Database (AMSD), or from the Crystallography Open Database (COD), for instance.
A file in the CIF format may contain the positions of atoms, as well as the symmetry group that the compound belongs to. As such, it is a very peculiar format: all atoms positions are not written explicitely in a CIF file; they have to be constructed by applying the appropriate symmetry operations. This is what Atomsk does when it reads a CIF file.
As an example, download the CIF file corresponding to the perovskite-type strontium titanate (SrTiO3), from the Website of the Crystallography Open Data Base:
This file is named "9002806.cif". If you open it with a text editor, you will see that it contains several informations:
As mentionned earlier, the CIF file does not contain the positions of all atoms in the unit cell. At least, not explicitely. Here, it contains only four atoms (Sr, Ti, and two oxygen atoms), while the chemical formula indicates "SrTiO3". However, the CIF file contains the space group and the symmetry operations relevant for this compound. By applying those symmetry operations to the atoms, it is possible to generate all equivalent positions, and fill the unit cell with atoms.
For the sake of example, let us convert this CIF file into the XSF format for visualization:
atomsk 9002806.cif vesta
You may notice that Atomsk indicates: "Applying symmetry operations". It is important to realize that, when reading a CIF file, Atomsk applies the symmetry operations immediately. In other words it finds all equivalent positions for atoms inside the box.
The previous command generates a file named "9002806.vesta", that contains all 20 atoms of the cell. Visualization with VESTA also confirms it:
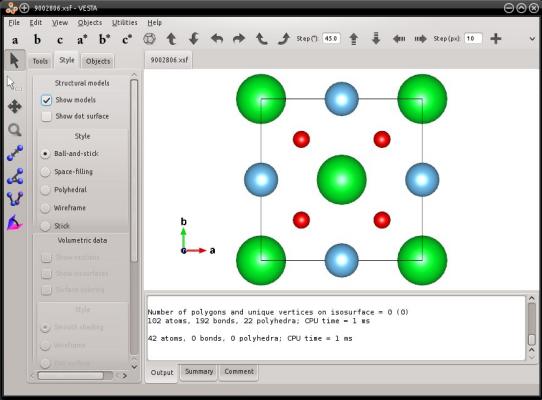
After reading the CIF file, Atomsk looses the information about the space group: all atom positions are known explicitely, and the space group is just P1. This is why the space group in the VESTA file is just "P1".
Sometimes, CIF files may specify partial occupancies. They mean that the same crystallographic site can be occupied by atoms of different species. Let us use another file from the COD as an example:
Ca0.35Sr0.65TiO3 CIF file from COD
This compound is similar to the previous one, except that this time, calcium atoms (Ca) can substitute strontium (Sr) atoms. If you open the CIF file ("9002807.cif"), you can see that the section about atoms contains an additional column, the "_atom_site_occupancy". All atoms have an occupancy of 1 (meaning that they occupy 100% of their site), except Ca and Sr atoms that have occupancies of 0.35 and 0.65, respectively. It is also important to note that Ca and Sr atoms are exactly at the same position.
Now let us read this file with Atomsk, and convert it into the VESTA format for visualization:
atomsk 9002807.cif vesta
In this example, Atomsk reads the information about partial occupancies from the CIF file, so it can write it into the VESTA file. Let us visualize it:
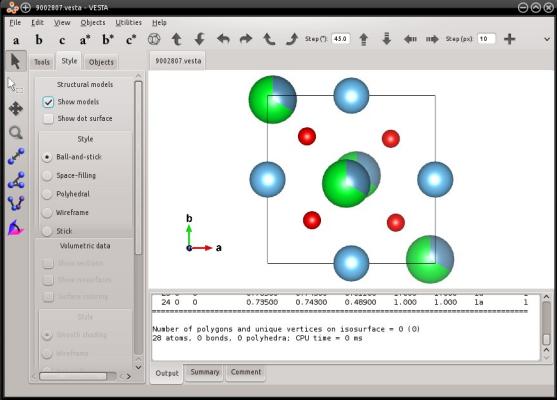
VESTA displays the partial occupancies as partially filled spheres, confirming that the information was conserved during the conversion.
Now, assume that we want to perform a simulation with LAMMPS. Let us convert the CIF file directly into a LAMMPS data file:
atomsk 9002807.cif lammps
In this case, Atomsk will display a warning message:
/!\ WARNING: data contains partial occupancies, which are
not supported by some output format(s).
Some atoms may overlap in the output file(s), which is not physical.
Indeed, it is not possible to write information about the partial occupancies into a LAMMPS data file, because this file format does not support it. So, this information is lost, and you end up with an atomic system where some atoms share exactly the same position, which is not physical. In such a case, it is up to you to decide what you want to do with overlapping atoms.
When reading CIF files, Atomsk saves the information about partial occupancies. Then, whenever possible, it writes it into output files if their format allows it. If not, Atomsk displays the warning above.
In Atomsk, the option "--create" allows to choose the orientation of a crystal when it is created. However this is not possible when an initial input file (like a CIF file) is read, therefore one has to use other methods to change the crystal orientation.
Let us use the CIF file for SrTiO3 above as an example (the file "9002806.cif"). First we have to determine the initial crystal orientation. It turns out that in this CIF file, the crystal is described with the orientation X=[110], Y=[110], Z=[001] (this can be deduced from the symmetries in that crystal).
Now, if we want to obtain another orientation, we have to rotate the crystal. Assume that we want X=[100], Y=[010], Z=[001]. Because this crystal is cubic, we can use the option "-orient", where the initial and final orientations must be provided:
atomsk 9002806.cif -orient [110] [-110] [001] [100] [010] [001] SrTiO3_rotated.cfg
Alternatively, it can be demonstrated that this corresponds to a rotation of 45° around the Z axis, which can be performed with the option "-rotate":
atomsk 9002806.cif -rotate Z 45 SrTiO3_rotated.cfg
Both commands yield the same result, as can be verified with OVITO:
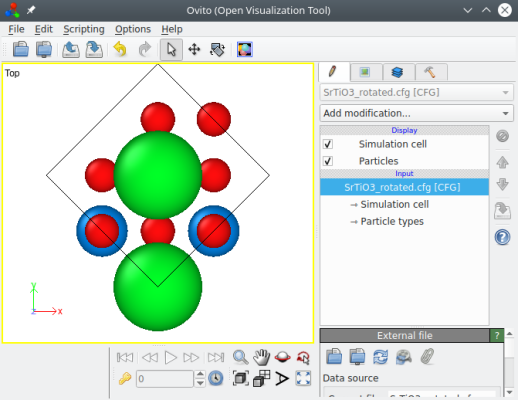
Now we have the correct crystal orientation, but the cell is rotated. Most of the time, we want an orthogonal cell with box vectors aligned with Cartesian axes. This can be achieved with the option "-orthogonal-cell":
atomsk 9002806.cif -rotate Z 45 -orthogonal-cell SrTiO3_rotated.cfg
This time the system is rotated, and we have a "proper" unit cell:
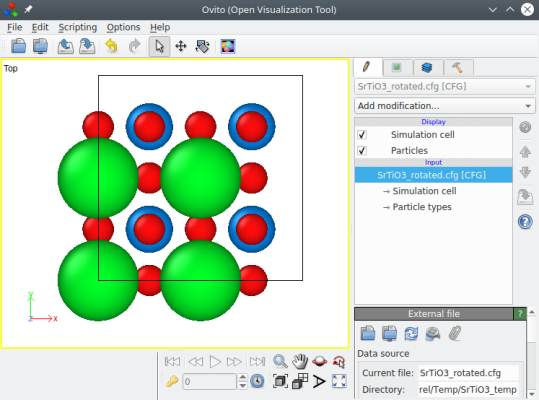
To summarize: in order to orient a unit cell that is read from a file (e.g. a CIF file), first determine the angle of rotation that you need, and then use the option "-orthogonal-cell".
Atomsk can only write very simplified CIF files. It always assumes that the space group is P1, and writes all atom positions into the CIF file.