Atomsk
Le couteau suisse pour les simulations atomiques

Le couteau suisse pour les simulations atomiques




Ce tutoriel explique comment calculer une fonction de distribution radiale (FDR) avec Atomsk.
▶ Pour plus d'information, référez-vous à la page de documentation correspondante.
ⓘ La construction de la liste de voisins est assez lente dans Atomsk jsuqu'à la version beta 0.11.2. Pour de meilleures performances, utilisez la version beta 0.12 ou ultérieure.
La fonction de distribution radiale ou FDR (en anglais, radial distribution function ou RDF), souvent notée g(r), est une mesure de la densité moyenne autour des atomes. Si vous n'êtes pas familier avec ce concept, vous pouvez lire la page de Wikipedia.
Commençons par l'exemple simple d'une maille élémentaire d'aluminium. Commençons par générer cette maille :
atomsk --create fcc 4.056 Al Al_unitcell.cfg
Ensuite, créons un fichier nommé "liste.txt" avec un éditeur de texte (comme Bloc-Notes, gedit, emacs, vi, ou similaire), et écrivons-y le nom du fichier à analyser, "Al_unitcell.cfg" :
Al_unitcell.cfg
Pour gagner du temps, vous pouvez générer le fichier "liste.txt" grâce à la commande suivante (sur les systèmes UNIX/Linux) :
ls Al_unitcell.cfg > liste.txt
Nous sommes maintenant prêts à calculer la FDR. Atomsk a besoin du nom du fichier contenant la liste, suivi de la distance maximale jusqu'à laquelle calculer la FDR, et la taille de l'échantillon. Par exemple, pour calculer la FDR jsuqu'à une distance de 12 Å, en utilisant un échantillonnage (ou "peau") de 0.1 Å:
atomsk --rdf liste.txt 12 0.1
ⓘ Au lieu de "liste.txt", vous pouvez appeler votre fichier comme vous le souhaitez. Assurez-vous de bien utiliser le même nom de fichier lorsque vous exécutez le mode "--rdf".
Atomsk écrit la FDR dans un fichier nommé "rdf_total.dat", que nous pouvons visualiser avec un logiciel de tracé de graphes comme xmgrace, gnuplot, ou logiciels similaires :
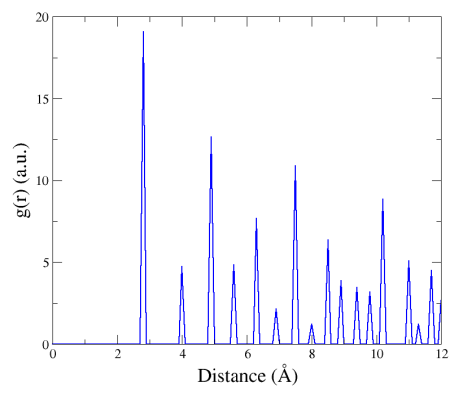
Ici, le premier pic à 2.86 Å correspond à la distance entre premiers voisins dans le réseau cfc (c'est-à-dire a0/√2), le second pic aux seconds voisins (a0), et ainsi de suite.
Notez que, bien que la maille élémentaire est très petite (un cube de 4.046 Å de côté), Atomsk est capable de calculer la FDR jusqu'à une distance beaucoup plus grande. C'est parce qu'il utilise toujours des conditions aux limites périodiques pour faire ce calcul, de sorte qu'un atome peut avoir ses propres répliques périodiques comme voisins.
Maintenant, construisons un système plus grand (super-cellule) et déplaçons les atomes de façon aléatoire :
atomsk Al_unitcell.cfg -duplicate 20 20 20 -disturb 0.2 Al.cfg
Écrivons le nom du fichier "Al.cfg" dans un fichier texte, puis exécutons Atomsk en mode "--rdf" :
ls Al.cfg > liste.txt
atomsk --rdf liste.txt 12 0.1 -wrap
ⓘ Puisque des atomes ont été déplacés, certains peuvent se retrouver en dehors de la boîte. Nous utilisons l'option "-wrap" pour s'assurer que tous les atomes soient bien remis dans la boîte avant le calcul de la FDR. Si vous omettez l'option "-wrap", alors Atomsk vous demandera si vous souhaitez l'appliquer avant de faire le calcul. Notez que si vous refusez, alors la liste de voisins risque d'être fausse, et donc la FDR risque aussi d'être erronnée.
Cette fois, la FDR ressemble à cela :
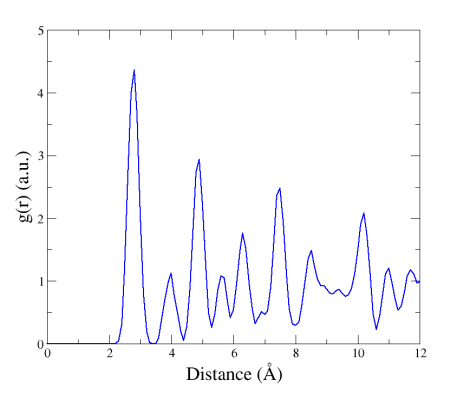
Nous pouvons voir que les pics sont élargis, parce que les atomes sont tous légèrement décalés par rapport à leur site cristallin. À cause de ce désordre, à longue distance la FDR tend vers une courbe constante et égale à 1.
Après une simulation en dynamique moléculaire (DM), il est parfois utile de calculer la FDR et de la moyenner sur plusieurs pas de temps. Réaliser une simulation de DM sort complètement du cadre de ce tutoriel, nous allons donc nous contenter de générer plusieurs configurations dans lesquelles les atomes sont déplacés aléatoirement, par exemple avec ces commandes :
atomsk Al_unitcell.cfg -duplicate 20 20 20 -disturb 0.2 Al_1.cfg
atomsk Al_unitcell.cfg -duplicate 20 20 20 -disturb 0.2 Al_2.cfg
atomsk Al_unitcell.cfg -duplicate 20 20 20 -disturb 0.2 Al_3.cfg
Nous allons prétendre que ces trois configurations sont des instantanés d'une même simulation en DM. Tout ce que nous avons à faire, c'est d'écrire les noms de ces fichiers dans un fichier-liste, et exécuter Atomsk en mode "--rdf":
ls Al_[1-3].cfg > liste.txt
atomsk --rdf liste.txt 12 0.1 -wrap
ⓘ Les options (comme l'option "-wrap" utilisée ici) sont appliquées à chaque système avant le calcul de sa FDR.
Atomsk va calculer la FDR pour chaque fichier, et ensuite calculer la moyenne sur toutes les configurations. Si ces configurations sont réellement des instantanés issus de la même simulation à des instants différents, alors vous obtiendrez la FDR moyennée dans le temps.
Les exemples ci-dessus ont utilisé l'aluminium comme exemple, qui ne contient qu'un seul type d'atomes. Maintenant, construisons un système contenant plusieurs types d'atomes, par exemple l'oxyde de magnésium MgO avec la structure "rock-salt" :
atomsk --create rocksalt 4.2 Mg O -duplicate 10 10 10 -disturb 0.2 MgO.cfg
Comme précédemment, écrivons le nom de ce fichier dans un fichier texte, et exécutons le mode "--rdf" :
ls MgO.cfg > liste.txt
atomsk --rdf liste.txt 12 0.1 -wrap
Vous pourrez remarquer que cette fois, Atomsk produit quatre fichiers, contenant les FDR partielles pour chaque paire possible de types d'atomes, ainsi que la FDR totale :
ⓘ Notez que la FDR pour les paires Mg-O est identique à la FDR pour les paires O-Mg, Atomsk ne calcule donc que la première et pas la seconde.
Vous pouvez visualiser ces graphes individuellement, ou bien les superposer sur le même graphe :
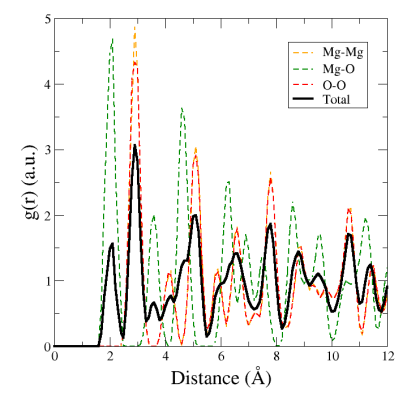
Le premier pic (tirets verts) correspond à la distance entre premiers voisins Mg-O. Le second pic (tirets oranges) correspond à la distance entre seconds voisins, qui est la même pour Mg-Mg et O-O. Ceci est typique du réseau cristallin NaCl ou "rock-salt". Ici encore, les pics sont relativement larges car nous avons déplacés les atomes aléatoirement avec l'option "-disturb".
À titre d'exercice, vous pouvez utiliser Atomsk pour générer une maille d'un matériau perovskite, qui contient trois types d'atomes différents, et ensuite calculer ses FDR. Vous verrez que là aussi, Atomsk générera une FDR pour chaque paire possible d'atomes, ainsi que la FDR totale.
Le calcul de FDR est très rapide pour les petits systèmes, comme dans les exemples ci-dessus. Pour des systèmes plus grands, il faut s'attendre à ce qu'elle soit plus lente, surtout parce que l'algorithme de recherche de voisins implémenté dans Atomsk aujourd'hui est O(N2). Autrement dit, le temps passé à rechercher des voisins augmente en fonction du carré du nombre d'atomes : doubler le nombre d'atomes multipliera ce temps par quatre. Par ailleurs, augmenter le rayon R, ou diminuer la taille d'échantillonnage dR, augmente aussi le temps de calcul des FDR.